


Targeting SRSF10 might inhibit M2 macrophage polarization and potentiate anti-PD-1 therapy in hepatocellular carcinoma
《Cancer Communications》(IF=20.0992)
摘要
背景:
免疫检查点封锁治疗肝细胞癌(HCC)患者的疗效仍然较差。虽然富含丝氨酸和精氨酸的剪接因子(SRSF)家族成员在肿瘤中发挥着重要作用,但它们对肿瘤免疫学的影响尚不清楚。本研究旨在阐明SRSF10在HCC免疫治疗中的作用。
方法:
为了确定与免疫治疗耐药性相关的关键基因,我们进行了单核RNA测序、多重免疫荧光分析、癌症基因组图谱和基因表达综合数据库分析。
我们利用体外共培养系统、流式细胞术、各种肿瘤荷瘤小鼠模型和患者来源的器官型肿瘤球状体,研究了SRSF10在免疫逃避中的生物学功能。
结果:
SRSF10在各种肿瘤中表达上调,并与预后不良相关。此外,SRSF10正向调控乳酸生成,SRSF10/糖酵解/组蛋白H3赖氨酸18乳酸化(H3K18la)在肿瘤细胞中形成一个正反馈回路。
乳酸水平升高促进M2巨噬细胞极化,从而抑制CD8+ T细胞活性。在机制上,SRSF10与MYB的3‘-非翻译区相互作用,增强MYB RNA的稳定性,随后上调关键的糖酵解相关酶,包括葡萄糖转运体1(GLUT1)、己糖激酶1(HK1)、乳酸脱氢酶A(LDHA),导致细胞内和细胞外乳酸水平升高。
乳酸积累诱导组蛋白乳酸化,进一步上调SRSF10的表达。此外,肿瘤产生的乳酸诱导组蛋白H3K18la位点在转运到巨噬细胞时发生乳酸化,从而激活转录并增强前肿瘤巨噬细胞的活性。
M2巨噬细胞反过来抑制了CD8+ T细胞的富集和干扰素-γ+CD8+T细胞在肿瘤微环境(TME)中的比例,从而产生了免疫抑制TME。在临床上,SRSF10可以作为评估各种实体肿瘤免疫治疗耐药性的生物标志物。
使用选择性抑制剂1C8对SRSF10的药理靶向作用增强了程序性细胞死亡1(PD-1)单克隆抗体(mAbs)在小鼠和人类临床前模型中的疗效。
结论:
SRSF10/MYB/糖酵解/乳酸轴对于触发免疫逃避和抗pd-1耐药性至关重要。通过1C8抑制SRSF10可能克服HCC中的抗pd-1耐受性。
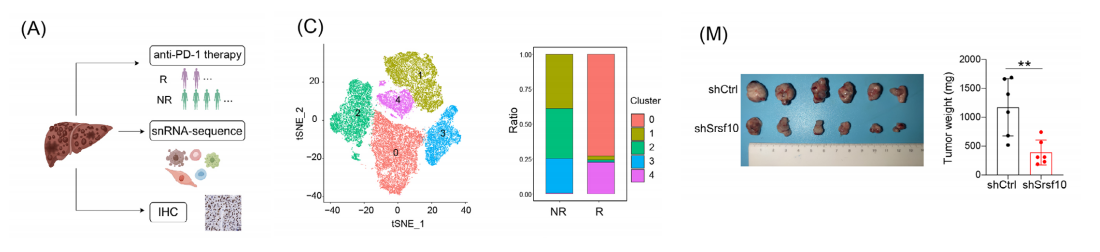
我们对6例接受抗pd-1治疗的患者进行了snRNA-seq检测,KEGG富集分析显示,集群2和集群3中的细胞在无应答患者中比在应答患者中更普遍,在与糖代谢相关的通路中富集。
此外,对经抗pd-1处理的批量RNA-seqHCC队列的富集分析,证实了无应答组中葡萄糖代谢通路的富集。
基于这些发现,我们推断了HCC中的葡萄糖代谢和免疫治疗耐药性之间的密切关系。将来自TCGA和GEO数据库的大量RNA-seq数据与预后信息进行整合,结果发现SRSF10是一个与糖酵解显著相关的基因。
我们检测了snRNA-seq队列中SRSF10在不同细胞类型中的表达,发现它主要在恶性细胞中表达。利用scRNA-seq数据集GSE149614,我们发现SRSF10主要作用于肿瘤细胞的现象。
随后,我们的研究结果强调了SRSF10在不同肿瘤类型中的普遍过表达,在RNA和蛋白水平上都很明显。SRSF10表达的增加与免疫抑制TME之间存在一致的关联。
我们生成了srsf10敲低的Hepa1-6细胞,并将它们原位移植以诱导原位HCC肿瘤。与对照组相比,shSrsf10组的原位肿瘤大小显著减少。
为了更深入地研究SRSF10在TME中的作用,我们对对照组和shSrsf10 HCC肿瘤进行了scRNA-seq检测。
结果明确表明,在shSrsf10治疗后,TME内的肿瘤细胞丰度显著下降。下调肿瘤中CD4+和CD8+ T细胞明显富集,而对照肿瘤中主要的巨噬细胞和单核细胞群相反。
这些发现表明,受SRSF10影响的巨噬细胞的改变主要涉及组织驻留的巨噬细胞。
这些确证分析显示,在srsf10敲低的HCC肿瘤中,CD8+ T细胞增多,而对照肿瘤显示F4/80+和CD206+巨噬细胞占优势。
我们使用Hepa1-6细胞建立皮下HCC肿瘤。我们观察到srsf10敲低的HCC肿瘤显示出肿瘤生长速度和重量的下降。
随后,将肿瘤组织酶消化成单细胞进行流式细胞仪分析,进一步证实了对对照和shSrsf10 HCC肿瘤进行scRNA-seq分析的结果。
因此,这些共同的结果表明,SRSF10在HCC肿瘤中形成抑制性TME方面发挥了关键作用,并提示其可能与抗pd-1免疫治疗的耐药性有关。
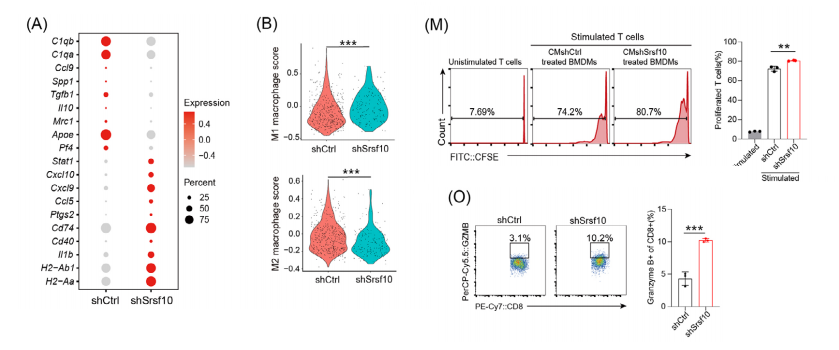
我们推测SRSF10通过调节M2巨噬细胞的极化,介导CD8+ T细胞对肿瘤细胞的细胞毒性杀伤作用。
我们利用对照组和shSrsf10 HCC肿瘤的scRNA-seq结果,分析了巨噬细胞中常见的巨噬细胞相关基因的表达。
在srsf10基因敲低小鼠的肿瘤中,肿瘤前巨噬细胞标记物的表达低于对照组小鼠的肿瘤,而抗肿瘤巨噬细胞标记物的表达则低于对照组小鼠在shSrsf10小鼠的肿瘤中高于对照组肿瘤,表明由shSrsf10介导的前肿瘤表型向抗肿瘤巨噬细胞表型的转变。
此外,shSrsf10肿瘤表现出M1巨噬细胞评分升高,而对照组肿瘤则倾向于更高的M2巨噬细胞评分。随后,用PMA处理THP1细胞,诱导向巨噬细胞分化,而从小鼠骨髓中分离骨髓细胞,用MCSF处理7天,获得BMDMs。
与对照组相比,与shSRSF10肿瘤细胞共培养的人和小鼠巨噬细胞中显著下调。在THP1细胞和bmdm细胞中,srsf10敲低的细胞中CD206显著下调。
通过趋化迁移实验,我们观察到shSRSF10 HCC细胞抑制了巨噬细胞的趋化募集。这些发现表明巨噬细胞在采用SRSF10介导的一种前肿瘤表型中发挥了重要作用,它们的缺失消除了SRSF10敲低对HCC肿瘤的抑制作用。
随后,我们的研究重点转向了CD8+ T细胞如何在srsf10敲低的HCC肿瘤中发挥作用。shSrsf10肿瘤中的效应CD8+ T细胞比对照组肿瘤中表现出更高的富集评分。
我们进行了细胞-细胞通信分析,发现在shSrsf10肿瘤中,巨噬细胞和CD8+ T细胞之间有更强的免疫促进趋化因子细胞间连接。
此外,来自shSrsf10肿瘤的CD8+ T细胞显示颗粒酶B(Gzmb)、Prf1、Tnf和Ifng的表达升高。CD8+ T细胞中Tcf7的表达增加。
为了研究CD8+ T细胞对SRSF10介导的免疫微环境的影响,我们进行了一个由肿瘤细胞上清液刺激的巨噬细胞和CD8+ T细胞的共培养实验。
这些结果表明,与与shSRSF10 HCC细胞共培养的巨噬细胞相比,与对照组HCC细胞共培养的巨噬细胞显著抑制了CD8+ T细胞的增殖和活化。
综上所述,SRSF10在肿瘤细胞中的表达通过促进巨噬细胞向具有前肿瘤表型的细胞转化,阻碍了CD8+ T细胞的细胞毒性功能。
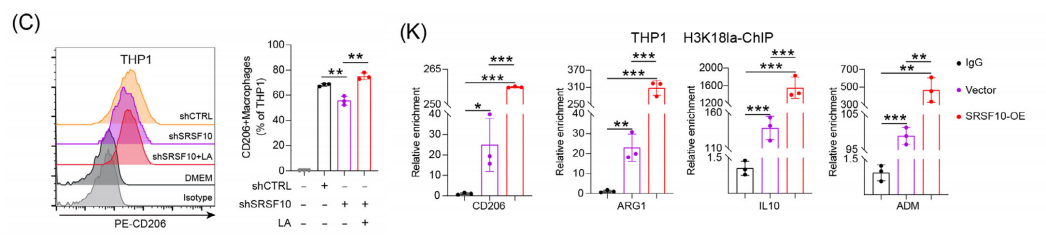
接下来,我们研究了SRSF10影响巨噬细胞极化的机制。将乳酸引入shSRSF10 HCC细胞来进行共培养实验。
PCR和流式细胞术检测显示,与shSRSF10肿瘤细胞共培养的巨噬细胞中M2巨噬细胞标记物的表达显著降低。然而,乳酸的加入逆转了这种下降。
与shSRSF10肿瘤细胞共培养后,巨噬细胞的迁移能力显著降低。然而,乳酸的添加有效地恢复了迁移能力。
我们推测乳酸通过组蛋白乳酸化促进M2巨噬细胞的极化。Westernblot分析显示,与过表达SRSF10的PLC/PRF/5细胞共培养的巨噬细胞中组蛋白乙酰化显著增加,而与shSRSF10 HCC细胞共培养的巨噬细胞中组蛋白乙酰化显著减少。
然而,补充乳酸(LA)恢复了它们的表达水平。我们的结果证实了H3K18la参与了srsf10介导的M2巨噬细胞极化。
组蛋白作为转录辅助因子在基因表达中起着至关重要的作用。ChIP检测显示,增加H3K18la修饰激活了M2巨噬细胞基因的转录表达,包括CD206、ARG1、IL10和ADM。
总之,我们提供的证据表明,肿瘤细胞中的SRSF10可以通过乳酸增强巨噬细胞的组蛋白乳酸化修饰,导致CD206等基因的激活,并促进免疫抑制TME。
为了研究SRSF10在糖酵解中的作用,我们对对照组和shSRSF10 Huh7细胞进行了批量RNA-seq检测,用DEGs进行了GSEA检测,显示糖酵解途径显著富集。
我们发现,在srsf10水平较高的患者中,与糖酵解相关的基因表达上调。为了研究SRSF10在肿瘤细胞代谢中的作用,我们进行了非靶向能量代谢测序。
我们鉴定了329个差异表达的代谢物,与糖酵解相关的代谢物在shSrsf10组中的丰度显著降低。此外,KEGG富集分析显示,碳代谢途径中的差异代谢物显著富集。
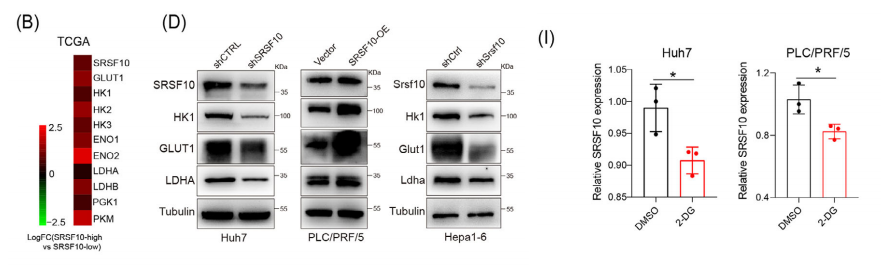
我们进一步检测了糖酵解相关基因(GLUT1、HK1和LDHA)在RNA和蛋白水平上的表达。在shSRSF10 Huh7和Hepa1-6细胞中,我们观察到这些基因的低表达。
相比之下,过表达SRSF10的PLC/PRF/5细胞显示这些糖酵解相关基因的表达升高。在过表达SRSF10和Hepa1-6细胞中ECAR和细胞内和细胞外的乳酸产量显著减少,PLC/PRF/5细胞中乳酸含量升高。
我们使用了MCT1和MCT4抑制剂。结果显示,BAY-8002抑制MCT1导致shSRSF10 HCC细胞的细胞外乳酸水平显著降低,而过表达SRSF10导致细胞外乳酸水平显著升高。
将对照组和shSRSF10 HCC细胞注射到小鼠肋骨下,产生皮下肿瘤。我们观察到,与对照组肿瘤相比,shSRSF10肿瘤中的乳酸产量显著减少。总之,我们发现SRSF10在体内和体外都正向调节肿瘤糖酵解和乳酸生成。
乳酸正向调控乳腺癌中SRSF10的表达。因此,我们研究了在HCC细胞中是否存在类似的机制。qPCR证实,补充LA可上调SRSF10的表达,而2-DG可下调SRSF10的表达。
Westernblot结果显示,LA随着泛kla和H3K18la水平的升高而增加了SRSF10的表达,而2-DG则相反。为了进一步了解乳酸调控SRSF10表达的具体机制,我们检测了其在组蛋白乳酸化中的作用。
我们假设乳酸增加了HCC细胞中的组蛋白乳酸化修饰,导致SRSF10的转录激活,如ChIP分析示,为涉及SRSF10/糖酵解/H3K18la轴的正反馈回路提供了证据。
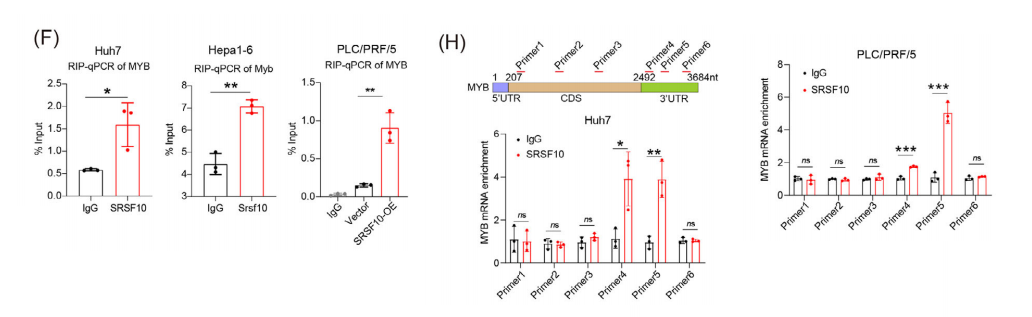
SRSF10主要通过rna结合活性发挥作用。RIP-seq实验KEGG富集分析显示,DEGs在与糖酵解相关的途径中富集,如癌症中的中心碳代谢。
RIP-seq和批量RNA-seq进行交叉,然后进一步与TCGA-LIHC数据集鉴定的与SRSF10高度相关的基因交叉。我们选择了与SRSF10相关性最强的MYB作为候选基因。
GSEA结果显示,与srsf10敲低组相比,对照组的MYB调控通路显著富集。PCR和Westernblot分析显示,SRSF10正向调控MYB的表达。
由于SRSF10是一个众所周知的剪接因子,我们首先进行了RIP分析,证实了SRSF10与MYB RNA的结合。我们将重点转移到探索SRSF10除剪接外调节RNA稳定性的机制上。
我们用放线菌素D处理肿瘤细胞。结果表明,在shSRSF10完成后,MYB RNA表现出更快的耗尽率,说明SRSF10在调节MYB RNA的稳定性中发挥了作用。
CLIP分析进一步显示,SRSF10主要结合在MYB的3‘非翻译区(3’UTR)区域。我们试图确定SRSF10的选择性抑制剂1C8是否可以调节MYB。
Westernblot分析显示,在Huh7和Hepa1-6细胞中加入1C8后,其迁移速率与CIP相似,表明1C8抑制了HCC细胞中SRSF10的活性。
综上所述,我们证明了SRSF10可以结合到MYB 3‘UTR区域来稳定其RNA,从而增加其蛋白表达。
接下来,我们证明了MYB在通过SRSF10调节糖酵解中的意义。我们的研究显示,与对照组细胞相比,MYB抑制后,HCC细胞中ECAR、乳酸水平和糖酵解相关基因显著降低。
此外,在shSRSF10 HCC细胞中,ECAR、乳酸水平和与糖酵解相关的基因显著下降,这可以被MYB逆转。此外,在用BAY-8002和AZD0095处理后,观察到对细胞外乳酸水平的不同影响。
这一发现表明,MYB上调诱导的细胞外乳酸水平的升高是由MCT4介导的。随后,我们发现,在干扰MYB的表达后,Huh7和Hepa1-6细胞的OCR显著增加,而ATP的产生显著减少,表明MYB驱动的糖酵解是主要的能量代谢途径。
在本研究中,我们研究了MYB调控糖酵解相关基因的机制。鉴于MYB是一个众所周知的转录因子,我们使用JASPAR数据库来预测了其与GLUT1、HK1和LDHA的转录结合序列。
ChIP检测证实,MYB转录上调了GLUT1、HK1和LDHA的表达。
总之,我们的实验表明,SRSF10与MYB RNA结合并增加其稳定性,从而激活与糖酵解相关的关键酶的转录表达,如GLUT1,并增加细胞内和细胞外的乳酸含量。
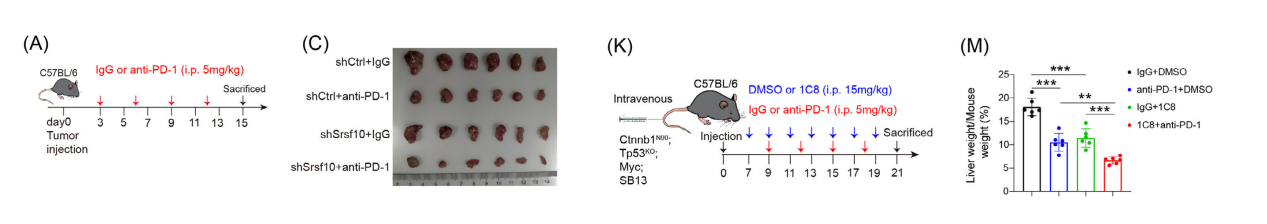
为了研究SRSF10缺陷是否会增强PD-1单抗治疗的效果,我们给接种shSrsf10或对照细胞的免疫活性小鼠注射PD-1单抗抗体或IgG同型对照。
与对照组相比,shSrsf10肿瘤治疗组或抗pd-1治疗组的肿瘤体积和重量显著减少。shSrsf10和抗pd-1联合使用对肿瘤体积和重量的抑制最为显著。
与单独使用shSrsf10或PD-1单抗治疗相比,shSrsf10联合PD-1单抗显著增加了肿瘤浸润性细胞毒性CD8+ T细胞数量,同时降低了F4/80+和CD206+巨噬细胞。
为了将我们的研究结果转化为抑制HCC生长的潜在治疗策略,我们将对照组和shSrsf10 Hepa1-6细胞皮下接种到野生型C57B/6小鼠中,并腹腔注射DMSO或1C8。
与对照组相比,shSRSF10组和shSRSF10+1C8组的肿瘤数量显著减少。我们观察到1C8显著减弱了肿瘤的生长和体重,但并不影响小鼠的体重。
此外,1c8处理的肿瘤中F4/80+和CD206+巨噬细胞的比例显著降低,而CD8+ T和IFN-γ+ CD8+ T细胞的比例增加。
为了更好地模拟HCC的TME,我们通过尾静脉注射诱导自发HCC肿瘤的形成。与对照组相比,单独使用1C8或抗pd-1治疗的小鼠的肿瘤负荷显著降低。
此外,1C8和抗pd-1联合治疗的小鼠的肿瘤负荷显著降低。生存分析显示,接受联合治疗的小鼠的OS时间延长。
流式细胞术实验证实,无论是1C8还是抗PD-1都能有效降低促肿瘤巨噬细胞的比例,增加了TME中CD8+ T细胞的比例。1C8和抗pd-1联合给药显示出更强的效果。
因此,在小鼠模型中,我们使用RNA干扰和药理抑制来证明SRSF10可能作为一种调节巨噬细胞群并恢复其抗肿瘤能力的策略。
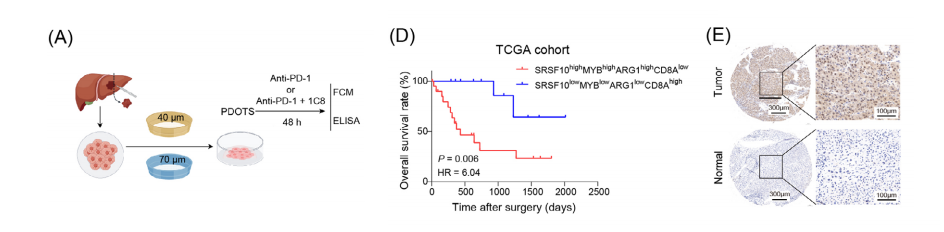
我们进行了进一步的研究,以确定1C8是否可以重塑人类的TME。从HCC患者中建立的患者源类器官PDOTS在DMEM中进行体外培养。
流式细胞术分析显示,1C8和抗pd-1联合治疗显著降低了肿瘤前CD206+巨噬细胞的比例,增加了CD8+ T细胞的比例。与单独使用抗pd-1相比,使用1C8和抗pd-1处理后,IFN-γ和PRF1的分泌显著增加。
因此,我们在人类临床前模型中的研究结果表明,用1C8靶向SRSF10是一种潜在的策略,可以调节巨噬细胞极化和恢复HCC患者的免疫杀伤TME。
我们的研究发现,SRSF10或MYB的低表达与HCC患者更良好的预后相关,SRSF10-MYB-ARG1高表达联合CD8轴低表达的患者的生存结果明显更差。
检测肿瘤组织中SRSF10与邻近正常组织中的表达情况。研究结果显示,SRSF10在肿瘤组织中的表达量较高。
SRSF10低表达表明HCC患者的预后明显优于高表达患者,这可以从TMA1队列中更长的OS和疾病生存期(DFS)得到证明。
接下来,我们收集了在中山医院接受抗pd-1免疫治疗的75例HCC患者的标本。IHC检测结果显示,对抗pd-1免疫治疗有反应的HCC患者的SRSF10的表达较低,而CD8的表达则较高。
为了进一步支持SRSF10与免疫治疗应答之间的联系,我们使用了三个公开的免疫治疗队列进行验证。
研究结果显示,SRSF10在无应答者中的表达高于有应答者。SRSF10表达较低的患者也表现出延长的OS。
这些数据共同证实了SRSF10的表达与使用抗pd-1治疗的肿瘤的免疫治疗反应和患者生存相关。
上述研究结果证实了在HCC中过表达的基因SRSF10是通过MYB促进糖酵解的关键作用。
此外,这说明SRSF10/糖酵解/H3K18la轴形成了一个正反馈回路,导致TME中糖酵解副产物乳酸积累。
随后,乳酸被运输到巨噬细胞中,在那里它通过乳酸化诱导组蛋白修饰。这种修饰进一步增强了肿瘤前巨噬细胞的极化,从而促进了免疫抑制的TME并导致PD-1免疫治疗耐药性。
我们的研究表明,SRSF10/MYB/糖酵解轴通过促进M2巨噬细胞极化来抑制TME。我们还发现,RNA干扰或对SRSF10的药理抑制重塑了TME,并增强了抗pd-1治疗的疗效。
此外,我们的研究显示,SRSF10抑制剂1C8在人源化肿瘤临床前模型中显示出了很好的抗肿瘤作用。因此,这些发现揭示了一种很有前途的抗pd-1联合治疗方案。

作者使用伯信生物明星产品CLIP试剂盒进行了上述筛选与分子互作调控机制的研究。
伯信好物推荐
CLIP Kit
产品介绍:
CLIP (crosslinking-immunprecipitation)是利用蛋白质和RNA在365 nm紫外光照射下会发生共价交联的特性,来研究蛋白质和RNA相互作用的重要技术。

伯信 CLIP Kit分为:
Bes3014-1 CLIP-qPCR Kit 12T
Bes3014-1 CLIP-qPCR Kit 30T
Bes3014-2 CLIP-seq Kit 12T
Bes3014-2 CLIP-seq Kit 30T
实验原理:
CLIP-qPCR(crosslinking-immunprecipitation and qPCR)即紫外交联免疫沉淀结合qPCR定量技术,通过免疫共沉淀RBP与RNA的结合复合物,获取消化(蛋白酶K、DNaseI、RNase T1)后的RNA,对RNA 3’端加接头,以及设计不同位点的引物,进行RNA结合蛋白位点的富集效率检测。
CLIP-seq(crosslinking-immunprecipitation and High throughput sequencing)即紫外交联免疫沉淀结合高通量测序技术,通过免疫共沉淀RBP与RNA的结合复合物,获取纯化及消化(蛋白酶 K、DNaseI、RNase T1)后的RNA,进行cDNA文库构建与测序分析。
技术流程:


结果实例:

产品优势:
1.经紫外照射,细胞内的RNA与相应的RNA结合蛋白交联,增强RNA与蛋白的结合能力
2.可检测多种RNA,如LncRNA、mRNA、microRNA
3. CLIP-qPCR可精确定位RNA与蛋白的结合位点
4.自主知识产权

