


Genome-wide enhancer RNA profling adds molecular links between genetic variation
and human cancers
《Military Medical Research》
(IF=21.1001)
背景:
增强子转录的失调发生在多种癌症中。增强子rna(eRNAs)是增强子的分裂产物,在转录控制中发挥关键作用。表征eRNA表达的遗传基础可能会阐明癌症的分子机制。
方法:
首先,在Cancer基因组图谱(TCGA)中对eRNA数量性状位点(eRNAQTLs)进行了综合分析,并利用多组学数据对其功能特征进行了表征。
为了建立中国结直肠癌(CRC)的eRNAQTL谱,我们利用表观基因组数据去除活性增强子,随后将其与154对CRC样本的转录和基因分型数据进行整合。
最后,我们进行了大规模的病例对照研究(34585例和69544例对照)和多管齐下的实验,以调查候选ernaqtl影响结直肠癌风险的潜在机制。
结果:
在30种不同的癌症类型中,共鉴定出300,112个eRNAqtl,它们通过调节染色质状态、与转录因子和rna结合蛋白结合来对eRNA转录发挥作用。
这些ernaqtl被发现在癌症风险位点中显著富集,这解释了癌症遗传性的一个重要原因。此外,肿瘤特异性ernaqtl对癌症的发展表现出较高的反应性。
此外,这些eRNAs的靶基因与癌症中信号通路失调和免疫细胞膨胀有关,突出了它们作为治疗靶点的潜力。
此外,多个民族人口研究认为,eRNAQTL rs3094296-T变异降低了中国人群(或=0.91,95%CI 0.88–0.95,P=2.92×10−7)和欧洲(或=0.92,95%CI 0.88–0.95,P=4.61×10−6)人群的结直肠癌风险。
在机制上,rs3094296对eRNA ENSR00000155786的转录具有等位基因特异性,作为转录激活因子促进其靶基因SENP7的表达。
这两个基因共同抑制了肿瘤细胞的增殖。我们整理的变异、基因和药物列表已经在CancereRNAQTL(http://canernaqtl.whu.edu.cn/#/)中提供,作为推进这一领域的信息资源。
结论:
我们的研究结果强调了ernaqtl在转录调控和疾病遗传性方面的重要性,并确定了基于erna的癌症治疗策略的潜力。

eRNAQTL分析最初是基于30种癌症类型的8757个个体的种系变异的基因型、eRNA表达和临床参数进行的。
随着样本量和eRNA数量的增加,检测到更多的eRNAQTLs,这意味着未来额外的RNA-seq数据集和增强子注释可能会揭示更多eRNAQtl。
与对照SNPs相比,ernaqtl在基因的上游和下游区域表现出明显的富集,而在基因间和内含子区域则表现出明显的缺失。
这些结果表明,已鉴定的ernaqtl高度富集增强子相关特征。
eRNA转录的起始受到染色质可及性和转录因子的调控。
因此,我们旨在探讨这些ernaqtl是否可以通过控制表观遗传学来调节eRNA的表达机制,利用编码项目的数据,观察到与增强子(H3K4me1、H3K27ac)和活性转录(H3K4me2、H3K79me2、H3K9ac和DNase I超敏位点)相关的组蛋白标记中eRNAQTLs显著富集,而抑制转录显著减少。
进行进一步的功能富集分析,发现有425个转录因子优先结合到eRNAQTLs周围的区域。
CCCTC结合因子(CTCF),一种参与调控3D基因组组织和转录调控的DNA结合因子,显示出广泛的富集。
在测试的150个RNA结合蛋白(rbp)中,发现有73个rbp表现出显著的富集,这表明它们可能在调节eRNA转录中发挥作用。
这些研究为表观基因组特征如何促进eRNA表达的调控提供了进一步的见解。
为了研究我们发现的ernaqtl是否能够深入了解目前无法解释的全基因组关联的机制,所有癌症相关snp都从GWAS目录中汇编。
随后,在与这些snp相关的连锁不平衡区域进行富集分析。我们进一步评估eRNAQtl是否可以作为疾病遗传力估计的补充。
基于LD评分回归的分区遗传力估计显示,ernaqtl约占CRC总遗传力的2-5%,与eqtl解释的遗传力相当。
这些研究表明,通过研究ernaqtl来探索分子机制可能为了解疾病易损性提供更多的见解。
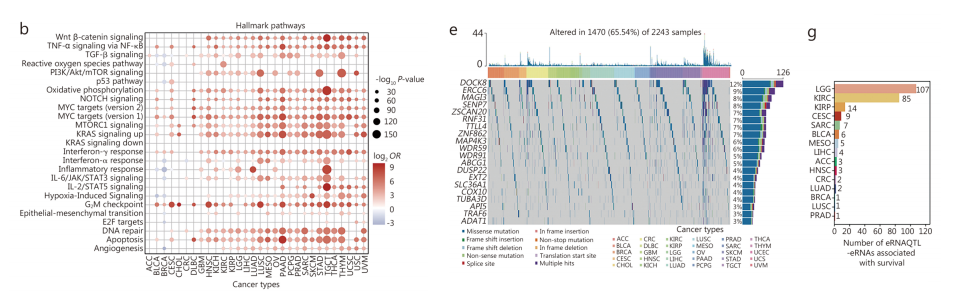
为了进一步阐明eRNAQTLl调控的eRNAs在人类癌症中的功能作用,我们首先检测了它们的表达模式。
有趣的是,共有982个的eRNAs只在一种癌症类型中表达,而297个eRNAs在≥10种癌症类型中广泛表达。
这些结果突出了不同癌症类型中erna的谱系特异性产生,这可能有助于对其生理功能的有价值的见解。
通过整合物理距离和共表达分析,实现了eRNAs靶基因的预测。共导致了14,670个相互作用,涉及6551个假定的靶基因和2132个erna。
此外通路分析显示,这些靶基因主要与转录调控和癌症免疫相关通路相关。为了研究这些靶基因所涉及的特异性途径,我们收集了50个标志性基因集和17个免疫相关基因集,进行基因集富集分析(GSEA)。
在来自20多种癌症类型的样本中,受eRNAs调控的基因被发现参与了典型的癌症途径,包括MYC、KRAS原癌基因(KRAS)和DNA修复途径。
此外,这些基因中平均有27.49%显示出细胞因子受体通路的活性,这被认为是免疫治疗的潜在靶点。
这些研究结果表明,eRNAs在致癌基因的调控以及细胞对缺氧或炎症等外部信号的反应中发挥着广泛的作用。
随后,我们使用肿瘤免疫估计资源估计了肿瘤膨胀免疫细胞的丰度,并确定了6个不同的亚群。
相关分析揭示了每种类型癌症中与免疫膨胀相关的100个eRNA调节因子的中位数,为确定癌症免疫调节的潜在机制提供了有价值的资源。
总的来说,erna靶点的识别增强了我们对其功能的理解,并阐明了增强子激活如何有助于肿瘤进展。
我们探讨了eRNA靶基因与癌症基因组事件相关的属性,发现这些基因,如胞质分裂奉献因子8(DOCK8)和切除修复6(ERCC6),表现出较高的突变和扩增或缺失变化负担。
从癌症药物敏感性基因组学(GDSC)数据库中共鉴定出194,660对erna-药物,并根据其药物靶点进一步分类。
研究发现,针对磷酸肌醇3-激酶/哺乳动物雷帕霉素靶蛋白(PI3K/mTOR)信号通路的药物与不同癌症类型中的多种eRNAQTL-eRNAs广泛相关。
这些结果表明PI3K/mTOR信号的过度激活通过调节细胞周期和增殖促进癌症转移和化疗耐药。
此外,我们还评估了eRNAQTL-eRNAs的临床相关性,并确定了250个与患者生存相关的eRNAs;然而,其中大多数是特异性的对于单个癌症类型,表明癌症模式。
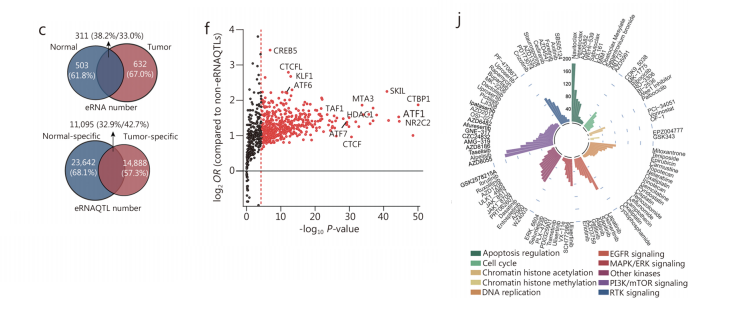
我们在中国CRC人群中进行了ATAC-seq、H3K27ac ChIP-seq、RNA-seq和高质量基因分型等多组学分析。
通过计算整合这些组织对的ATAC-seq和H3K27ac ChIP-seq的数据,肿瘤组织中平均有35340个增强子,成对正常组织中的27798个增强子。
将154个CRC肿瘤/邻近正常样本的RNA-seq reads进一步与增强子区域重叠,具有相对高表达水平的eRNAs被认为是可检测到的eRNAs,结果在肿瘤组织中发现943个eRNAs,在邻近正常组织中发现814个eRNAs。
然后,我们建立了154对CRC及邻近正常组织的frst全基因组eRNAQTL图谱,以揭示eRNAQTL在中国人群中的重要生物学功能。
与泛癌分析的结果一致,我们的肿瘤组织中的ernaqtl主要定位于增强子相关的功能区域。此外,在455个TFs和84个rbp中均观察到eRNAQTLs的显著富集。
有趣的是,TFBS中CTBP1、NR2C2和ATF1显著富集,这是CRC的关键驱动因素。为了阐明eRNAs在癌症发展中的分子功能,我们整合了来自154个CRC组织的eRNAs和mrna的表达数据,并进行了共表达分析,以确定eRNAs的候选靶基因。
此外,受erna调控的基因分别为涉及典型的癌症途径,包括内吞作用、轴突引导和抗菌肽抗菌肽(cAMP),表明有癌基因调控的趋势。
此外,我们发现有267个eRNAQTL-eRNAs与大量的免疫细胞膨胀有关。使用GDSC药物数据集共鉴定出7087对erna-药物对,其中针对PI3K/mTOR信号通路的药物排名最高。
158个eRNAQTL-eRNAs与AZD8055相连,这是一种靶向PI3K/mTOR信号通路的抑制剂。
我们对这些erna关于免疫细胞膨胀和药物靶点的评估重新证实了它们在临床应用中的潜力。.
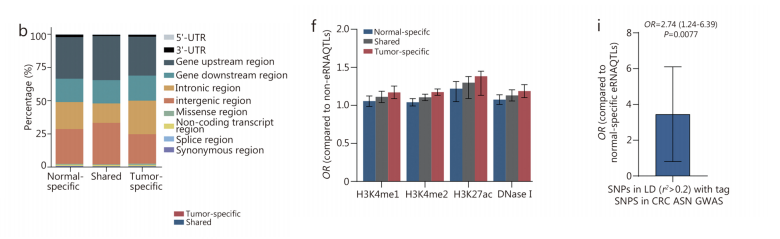
通过对肿瘤和正常状态的比较QTL分析来识别肿瘤特异性变异,是解释癌症发展的生物学机制的重要补充。
57.3%的肿瘤eRNAQTLs和68.1%的正常eRNAQTLs似乎具有组织特异性,而有11,095个共享的eRNAQTLs。
对eRNAQTL的缺陷大小的分析显示,在肿瘤发生后,共享的eRNAQTL缺陷的方向保持不变。此外,状态特异性和共享ernaqtl的功能分布也观察到类似的模式。
我们检测了TFs结合区域的富集,17个转录因子在肿瘤特异性ernaqtl中强烈富集,所有这些都在CRC中也显著上调。
相应地,肿瘤特异性eRNAQTL富集率与eRNAQTL共享富集率(TF)呈显著正相关,表明调控开关可能解释了一些肿瘤特异性eRNAQTL缺陷。
肿瘤特异性ernaqtl在活性组蛋白修饰中具有更强的富集作用。此外,曼哈顿图显示了三种类型的ernaqtl的不同分布模式,显著的位点位于不同的染色体上,促使我们进一步研究它们在CRC中的各种功能。
与正常特异性患者相比,肿瘤特异性患者中,亚洲血统CRC患者的肿瘤特异性ernaqtl富集,表明肿瘤特异性ernaqtl对癌症遗传性的贡献更大。
总之,这些结果揭示了特定肿瘤状态相关的ernaqtl所发挥的某些激活的功能作用可能有助于CRC肿瘤的发生。
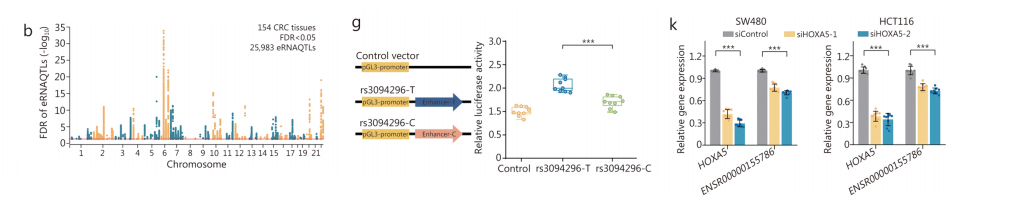
为了验证eRNA作为遗传和癌症风险之间的关键联系的作用,我们进行了涉及eRNaqtl的全面分析、一项大规模人群研究和一项CRC组织功能实验。
对来自中国的4293例病例和7176例对照进行了大规模的全基因组分析。有趣的是,在强LD中有11个snp位于3q12.3区域,这是与CRC风险相关的最显著的位点。在一项欧洲人群研究中也发现了该位点。
我们使用生物信息学工具对该LD块中的ernaqtl进行了功能注释。我们发现,eRNAQTL变体rs3094296在经典增强子特征和活性组蛋白修饰峰中富集。
因此,我们选择了rs3094296作为后续验证的候选对象。复制阶段I eRNAQTL rs3094296的T等位基因降低了结直肠癌的风险。在复制阶段II中进一步验证了这种关联。
最后,通过综合分析数据的发现和复制阶段,提出了令人信服的证据之间rs3094296和CRC风险在中国和欧洲人口。
接下来,我们研究了eRNAQTL rs3094296对CRC风险的贡献的潜在分子机制。eRNAQTL rs3094296与肿瘤组织中eRNA ENSR00000155786的表达显著相关,而在正常组织中无明显相关性。
随后,我们构建了一个含有不同等位基因的荧光素酶载体来评估增强子的活性。
结果表明,在HCT116和SW480细胞系中,rs3094296-T等位基因组的荧光素酶活性高于rs3094296-C等位基因,而在HIEC-6细胞中差异无统计学意义,表明eRNAQTL rs3094296调控eRNA ENSR00000155786的表达。
eRNAs的转录主要受TF与增强子元件结合的调控。同时,上述分析也显示TFBSs在eRNAQTLs中显著富集。
我们使用JASPAR进行了TF基序分析,并发现HOXA5基序与rs3094296-T等位基因特异性结合。
此外,HOXA5的敲除显著降低了rs3094296- T等位基因的增强子活性SW480细胞系,但对HIEC-6细胞无缺陷,表明肿瘤状态下HOXA5在肿瘤状态下优先结合rs3094296-T等位基因。
此外,我们观察到,在我们的CRC样本和TCGA CRC样本中,HOXA5与ENSR000000155786的表达呈正相关呈正相关。
此外,在SW480和HCT116细胞系中敲除HOXA5导致ENSR000000155786的表达降低,而在HIEC-6细胞中没有明显的差异。
这些结果共同表明,eRNAQTL rs3094296对eRNA ENSR00000155786表达的等位基因特异性差异是由HOXA5介导的。
为了阐明eRNA在癌症发展中的分子功能,我们进行了共表达分析,分析了由eRNA ENSR00000155786调控的下游靶基因。
ENSR000000155786和SENP7的表达水平的表达显著相关。在两个队列中,eRNAQTL rs3094296与SENP7的表达之间也存在正相关关系。
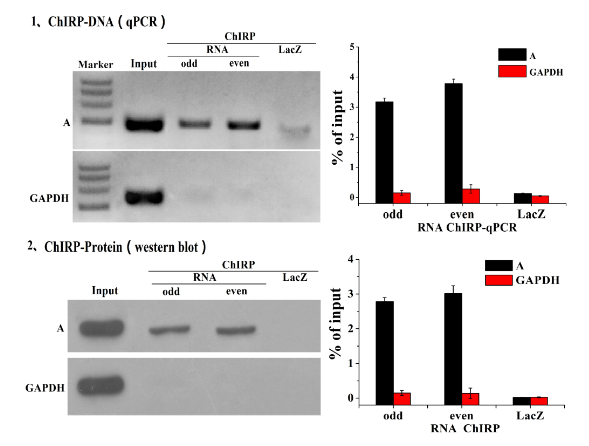
此外,ChIRP分析显示,与LacZ探针组相比,两个探针组的ENSR00000155786在SENP7的活性增强子和启动子区域均显著富集。
针对ENSR00000155786的敲除实验显示,SENP7的mRNA和蛋白水平均显著下降。同样,HOXA5的敲除也导致了CRC细胞系中SENP7表达的显著减少,但对HIEC-6细胞没有明显的缺陷。
综上所述,这些结果为eRNA ENSR00000155786作为转录激活因子调控邻近基因SENP7的表达提供了证据。
随后,我们探讨了ENSR00000155786和SENP7在结直肠癌发病机制中的功能作用。与正常组织相比,肿瘤组织表现出ENSR00000155786和SENP7的显著下调.此外,敲除ENSR000000155786或SENP7均显著提高了SW480、HCT116和HIEC-6细胞的增殖率。
有趣的是,这两个基因在细胞增殖上表现出协同效应。COLO205 CRC细胞系的全基因组CRISPR/Cas9功能缺失筛选数据证实了SENP7在细胞增殖中的重要作用。
综上所述,ENSR00000155786和SENP7在CRC肿瘤发生中具有潜在的肿瘤抑制基因,有望作为治疗靶点。
为了促进对eRNAQTL功能的探索的广泛研究,我们开发了一个用户友好的数据库,称为CancereRNAQTL 。用户可以对全面查询进行“单次搜索”和“批量搜索”。
在eRNAQTL模块中,用户可以根据癌症类型、SNP ID和特异性ifceRNA探索eRNAQTL结果。生存-eRNAQTL模块显示了与患者生存相关的eRNAQTL。
我们嵌入了箱形图来说明每个记录的eRNAQTL基因型和eRNA表达之间的关联。此外,我们还利用KM图来描述生存相关性。
扩展页面提供了一个eRNAQTL的列表,这些列表可以在与现有的eQTL结果合并时影响mRNA的表达。
CancereRNAQLT数据库还包含了已识别的eRNAs的靶基因、临床相关性和药物靶点,这些数据来自多组学数据进行分析,如转录组信息、临床数据集和药物基因组学数据。
总之,CancereRNAQTL是基因和癌症研究人员的全方位资源。
我们对30种不同癌症类型的eRNA表达的遗传效应进行了系统分析,从而创建了一个全面的eRNAQTL资源,有助于解释与癌症相关的合理变异。
我们的深入描述强调了ernaqtl及其靶eRNAs在肿瘤发生和临床应用中的重要性。重要的是,我们定量分析了中国患者CRC组织中增强子区域的表达,并建立了中国人的eRNAQTL谱。
RNAQTL rs3094296和CRC风险,通过大规模、多种族人口研究,包括34,585例病例和69,544例对照。
在机制上,该变异在促进eRNA ENSR00000155786和mRNA SENP7的转录方面表现出等位基因特异性,从而协同抑制肿瘤细胞增殖。
我们的研究揭示了注入erna表达的遗传变异进一步提高了我们对转录调控机制的理解,并为疾病的病因学提供了额外的见解。
 作者使用伯信生物明星产品CHIRP试剂盒进行了上述筛选与分子互作调控机制的研究。
作者使用伯信生物明星产品CHIRP试剂盒进行了上述筛选与分子互作调控机制的研究。
伯信好物推荐
CHIRP Kit
产品介绍:
ChIRP(Chromatin Isolation by RNA Purification)是一项研究RNA与DNA及蛋白质之间相互作用的技术。根据研究对象不同,ChIRP技术可分别结合高通量测序(ChIRP-Seq)和质谱技术(ChIRP-MS),研究与目标RNA互作的基因和蛋白质。
伯信CHIRP Kit 分为:
Bes5104-1(S) CHIRP-DNA Kit 12T
Bes5104-1(N) CHIRP-DNA Kit 30T
Bes5104-2(S) CHIRP-Protein Kit 12T
Bes5104-2(N) CHIRP-Protein Kit 30T
Bes5104-3(S) CHIRP-DNA、Protein Kit 12T
Bes5104-3(N) CHIRP-DNA、Protein Kit 30T
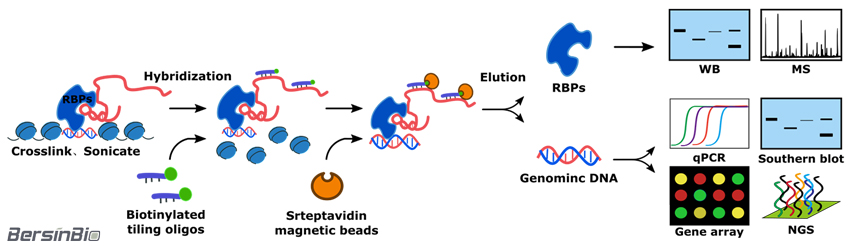
实验原理:
ChIRP方法通过设计生物素标记RNA互补探针,并使其与链霉亲和素结合,这样,在探针与目标RNA特异性结合的同时,捕获RNA结合调控的DNA染色体片段与参与转录调控的RNA结合蛋白质(RBPs)。
DNA染色体片段经过文库构建与高通量测序,在基因组水平上获得转录调控RNA(增强子RNA, eRNA)调控的下游靶基因,结合qPCR可以研究结合调控强度; RNA结合蛋白经过酶消化与高效液相-质谱分析,可以鉴定参与转录调控的蛋白质,结合Western Blot可以进一步研究结合作用强度。
技术流程:
结果实例:
产品优势:
1. 伯信独立研发,具有自主知识产权。
2. 特异性和灵敏度高,稳定性好。
3. 检测方法领先,结果准确、重复性好。
4. 快速检测,操作简单,安全便捷。

