


GLIS2 Prevents Hepatic Fibrosis by Competitively Binding HDAC3 to Inhibit Hepatic Stellate Cell Activation《Cellular and Molecular Gastroenterology and Hepatology 》(IF=8.797)

背景:
GLIS2在纤维化疾病中的作用存在争议。
据报道,GLIS2缺失有助于小鼠肾纤维化,并且还被报道可预防高脂诱导的小鼠肝纤维化。
方法:
采用CCL4诱导小鼠肝纤维化。
采用苏木精和伊红、马松、天狼星红和酶联免疫吸附法检测和评估人或小鼠肝纤维化的分期。
构建了四环素应答性GLIS2敲除肝星状细胞(HSCs)的研究模型,并命名为GLIS2-SG-Dox。
通过添加转化生长因子β1来刺激HSCs转分化,从细胞增殖、迁移、脂滴数量等方面综合评价HSCs的活化状态。
在机制研究中,采用双荧光素酶、共免疫沉淀、酵母双杂交系统、染色质免疫沉淀和DNA pulldown来研究或证明GLIS2参与调节肝纤维化的分子机制。
在整个研究中,实时荧光聚合酶链反应(定量逆转录聚合酶链反应)用于检测各靶基因信使RNA表达的相对丰度,Western blot用于检测蛋白质的相对丰度,免疫组化或免疫荧光用于观察目标蛋白的亚细胞定位。
结果:
GLIS2在人肝纤维化组织和CCL4诱导的小鼠肝纤维化组织中的表达明显降低,特别是在HSCs中。
在GLIS2-SG-Dox细胞中,过氧化物酶体增殖物激活受体γ (PPAR-γ)通路失活,细胞经历了肌成纤维细胞转分化转化。
过表达GLIS2可提高PPAR-γ的乙酰化水平,减轻CCL4诱导的小鼠肝纤维化。
从机制上讲,相对丰富的GLIS2和组蛋白去乙酰化酶3(HDAC3)形成螯合物,避免PPAR-γ的去乙酰化,从而维持 HSCs中PPAR-γ信号通路的激活水平。
在这个过程中,HDAC3作为GLIS2影响PPAR-γ信号传导的介质。
然而,当GLIS2缺失时,HDAC3使PPAR-γ去乙酰化,激活HSCs,导致肝纤维化。
结论:
GLIS2 缺陷促进 HSCs 的肌成纤维细胞转分化和活化。
在机制上,GLIS2 通过竞争性结合 HSCs 中的 HDAC3 来调节 PPAR-γ 的乙酰化。

对疑似肝癌纤维化患者的肝活检标本进行病理检查,结果显示,在纤维化加重的肝组织中,胶原蛋白升高,而GLIS2明显降低。
然后用CCL4诱导法建立肝纤维化小鼠模型。
从模型组和对照组的肝脏中分离出肝细胞和HSCs,发现GLIS2明显下降,尤其是在HSCs中。
因此,研究团队用HSCs来研究GLIS2对肝纤维化的影响。
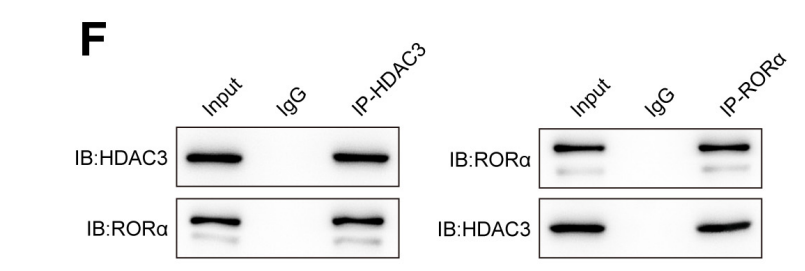
Co-IP和酵母双杂交系统测试证实了GLIS2和HDAC3,以及HDAC3和PPAR-γ的直接相互作用。
为了阐明调控机制,研究团队利用DNA pulldown等方法研究其调控通路和靶点。

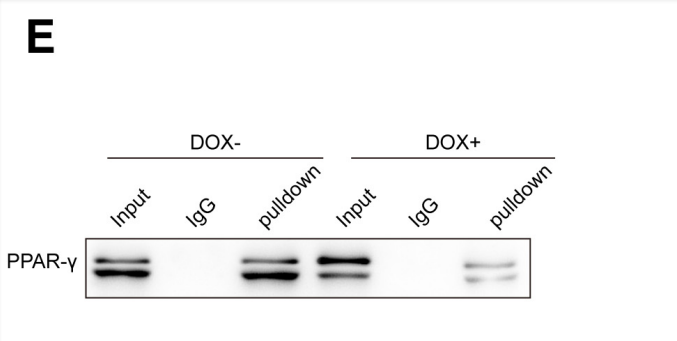
DNA pulldown实验显示了与CD36启动子结合的PPAR-γ的存在。
DNA pulldown实验发现GLIS2-SG细胞中HDAC3-RORa与CD36启动子结合。
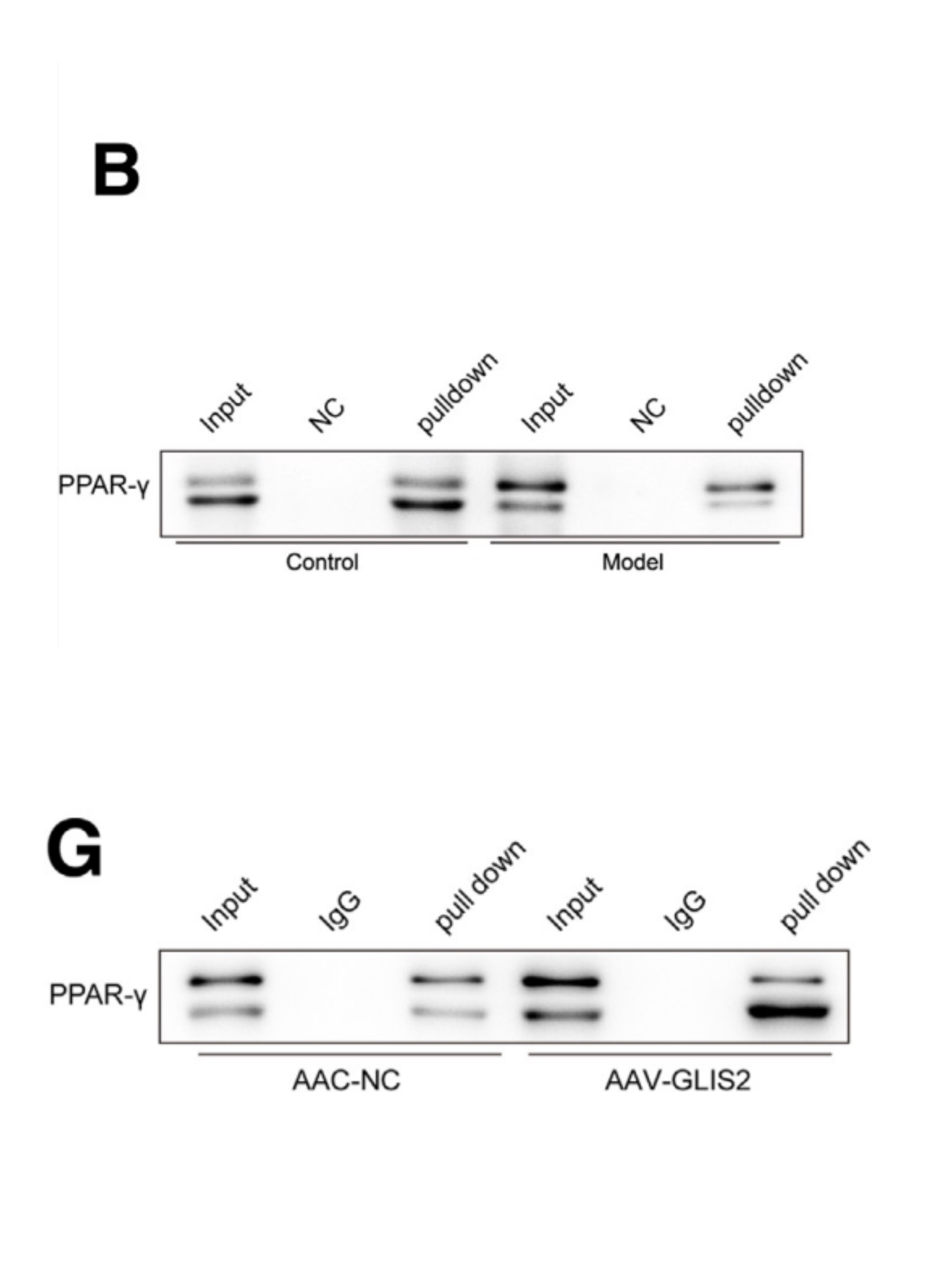
DNA pulldown实验发现,PPAR-γ可以在CCL4诱导的肝纤维化小鼠的HSCs中与CD36启动子结合。
在AAV-NC小鼠和AAC-GLIS2小鼠中,DNA pulldown实验检测到PPAR-γ与CD36启动子结合在HSCs中。
GLIS2通过与HDAC3的竞争结合来调节PPAR-γ的乙酰化,GLIS2的过表达确保了PPAR-γ的乙酰化水平,并减轻了小鼠的肝纤维化。
研究团队提出了一种激活HSCs的新机制: GLIS2缺失导致HDAC3过度结合PPAR-γ和PPAR-γ失活,从而抑制靶基因的转录激活。
此时,脂肪细胞表型转化为成肌细胞表型,导致HSCs的激活。
这些发现增加了对肝纤维化发病机制和预防的理解。
作者使用伯信生物明星产品DNA Pulldown试剂盒进行了上述分子互作调控机制的研究。
原文链接:https://www.cmghjournal.org/article/S2352-345X(22)00226-0/fulltext

