


N6-methyladenosine-modifed circRIMS2 mediates synaptic and memory impairments by activating GluN2B ubiquitination in Alzheimer’s disease
背景
突触退化发生在阿尔茨海默病(AD)的早期阶段,在毁灭性的症状之前,与认知能力下降密切相关。环状rna(环状rna)在神经组织中大量丰富,环状rna的异常表达先于AD症状,与临床痴呆的严重程度显著相关。然而,AD早期环状rna失调与突触损伤之间的直接关系尚不清楚。
方法
通过海马全转录组测序来鉴定4个月大的野生型和APP/PS1小鼠的失调环状rna和miRNAs。利用RNA反义纯化和质谱技术揭示了circRIMS2与甲基转移酶3、n6-腺苷-甲基转移酶复合物催化亚基(METTL3)之间的相互作用。circRIMS2/miR-3968在突触靶向ube2k介导的NMDA受体GluN2B亚基泛素化中的作用通过许多慢病毒,随后通过形态学染色、共免疫共沉淀和行为测试进行评估。此外,我们还使用了一种膜透性肽来阻断AD小鼠中K1082在GluN2B上的泛素化。
结果
circRIMS2在4个月大的APP/PS1小鼠中显著上调,这是由mettl3依赖的n6-甲基腺苷(m6A)修饰介导的。过表达circRIMS2导致4月龄C57BL/6小鼠的突触和记忆障碍。MiR-3968/UBE2K被验证为circRIMS2的下游。UBE2K水平的升高可通过泛素化GluN2B上的K1082来诱导AD的突触功能障碍。沉默METTL3或阻断K1082在GluN2B上的泛素化,可以显著挽救AD小鼠的突触功能障碍。
结论
总之,我们的研究表明,m6a修饰的circRIMS2通过海绵化miR-3968激活ube2k依赖的GluN2B泛素化和降解,介导AD的突触和记忆损伤,为AD提供了新的治疗策略。
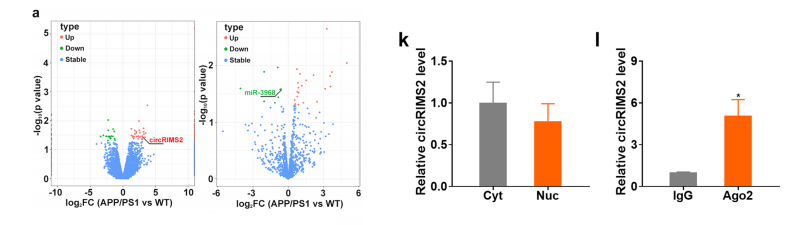
在AD早期和前-痴呆症症状。我们进行了RNA测序鉴定,qRT-PCR验证了APP/ PS1小鼠中circRIMS2/miR-3968 ceRNA成对的上调,且circRIMS2的上调和下调miR-3968。在3×Tg小鼠和Aβ处理的初级皮质小鼠神经元中,我们观察到circRIMS2/miR-3968类似的变化,表明该通路在几种AD模型中被异常激活。
FISH鉴定了circRIMS2和miR-3968在N2a细胞的细胞质中的共定位。双荧光素酶报告基因检测和qRT-PCR结果显示,circRIMS2抑制了miR-3968的表达,表明其miRNA海绵潜能。actinomycin D用于阻断新的RNA合成,circRIMS2比线性RIMS2具有更高的稳定性。
circRIMS2在细胞内的分布在N2a细胞的细胞核和细胞质之间没有明显的差异。我们发现circRIMS2被Ago2显著富集,表明其miRNA海绵潜力。
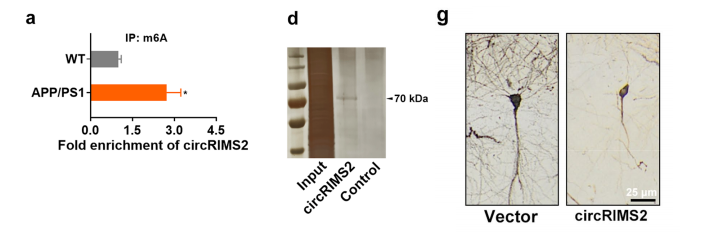
在4个月大的APP/PS1小鼠中,circRIMS2的Te m6A水平显著上调。
RNA下拉和质谱分析认为METTL3是一个与circrims2结合的m6A调节因子.过表达METTL3增加了circRIMS2的m6A水平。敲除METTL3逆转了Aβ诱导的N2a细胞中circRIMS2的m6A修饰,并降低了Nc2a细胞中ActcRIMS2的稳定性。
这些结果表明,mettl3介导的m6A修饰稳定了circRIMS2。
为了说明circRIMS2在AD发病机制中的作用,我们将一种含有circRIMS2的慢病毒注射到4月龄C57BL/6小鼠的海马中。过表达circRIMS2的小鼠在学习阶段表现出更长的潜伏期。高尔基体染色观察到树突棘密度和蘑菇型棘的百分比的降低。circRIMS2的过表达会降低突触蛋白的蛋白水平,包括GluN2B和GluN2A。
此外,我们观察到circRIMS2可以海绵化miR-3968,并导致体内miR-3968的减少。这些结果表明,circRIMS2的过表达诱导了体内的记忆和突触损伤。
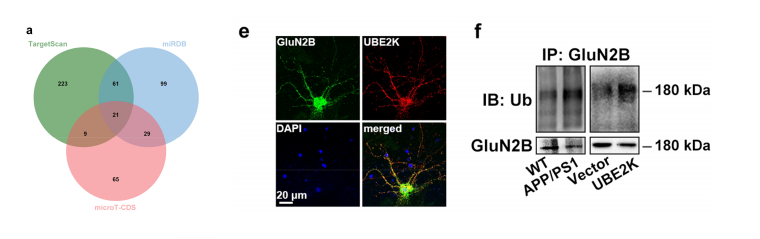
我们运用3个软件预测miR-3968的靶点,它介导AD的突触功能障碍,均发现了21个基因,这些基因在蛋白泛素化GO项中富集。我们选择了TRIM62、RC3H1和泛素结合酶E2 K(UBE2K)作为进一步验证的候选酶。实验表明,在过表达mir-3968的N2a细胞中,只有UBE2K蛋白水平显著下调,表明UBE2K是miR-3968的潜在靶点。
我们进行了荧光素酶活性测定,发现在HEK293细胞中,miR-3968只降低了WT UBE2K的荧光素酶活性,而没有降低UBE2K3‘UTR的两个突变体。我们观察到,上调miR-3968或沉默UBE2K可以挽救过表达circRIMS2的小鼠的记忆和突触损伤。
我们进一步研究了UBE2K介导AD突触损伤的下游靶点,发现GluN2B可能是NMDA受体的一个亚基,可能是神经元通信[27]的重要靶点。研究表明,UBE2K可能直接与GluN2B相互作用,介导其泛素化和降解,因为过表达UBE2K降低了GluN2B蛋白水平,而UBE2K沉默增加了GluN2B蛋白水平。
在C57BL/6小鼠中,GluN2B抗体也可以降低UBE2K。此外,在APP/PS1小鼠和ube2k转染的N2a细胞中,GluN2B蛋白水平降低,而泛素化水平升高。我们使用GPS-Uber预测了GluN2B中潜在的泛素化位点,发现了fve高度保守的泛素化位点。荧光素酶活性测定提示GluN2B可能不是miR-3968的直接靶点。
这些数据表明,circRIMS2/ miR-3968通路的上调可能通过K1082位点的GluN2B泛素化导致UBE2K的异常激活和GluN2B的降解。
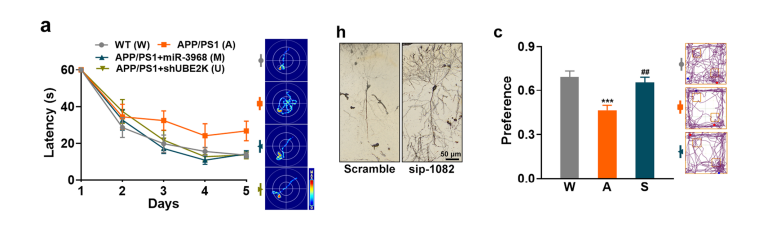
我们研究了下调circRIMS2/miR-3968/UBE2K通路是否可以挽救AD患者的突触和记忆损伤。通过上调miR- 3968或沉默UBE2K来下调该通路。含有miR-3968或shUBE2K的慢病毒注入APP/ PS1小鼠的海马中,部分挽救了记忆损伤,逆转了APP/PS1小鼠突触蛋白的蛋白水平,表明下调circRIMS2/miR- 3968/UBE2K通路显著挽救了AD的突触和记忆损伤。
我们生成了两个分别重叠于K1082和K1097泛素化位点的多肽段。sip-1082可以逆转N2a细胞中过表达UBE2K所引起的GluN2B蛋白水平的下降。表明只有K1082R突变而不是K1097R突变消除了UBE2K与GluN2B-2之间的相互作用。
我们给药sip-1082在12个月大的APP/PS1小鼠。在NOR和MWM测试中,处理显著提高了其学习能力,恢复了神经元树突的密度、成熟和复杂性,抑制了GluN2B泛素化,增加了GluN2B蛋白水平。结果表明,阻断ube2k介导的GluN2B泛素化可以显著挽救AD小鼠的突触和记忆损伤。阻断circRIMS2/ miR-3968/UBE2K/GluN2B轴可显著改善APP/PS1小鼠的突触功能障碍。
我们还应用MeRIP-PCR检测N2a细胞中转染METTL3后UBE2K和GluN2B的m6A水平,结果显示METTL3未染UBE2K和GluN2B的m6A水平,部分排除了METTL3对UBE2K和GluN2B的直接m6A修饰调控。在MWM和NOR测试中,沉默METTL3部分挽救了记忆障碍,逆转了APP/PS1小鼠的突触蛋白水平以及circRIMS2的表达和m6A水平,表明下调METTL3可显著减轻APP/PS1小鼠的AD病理。
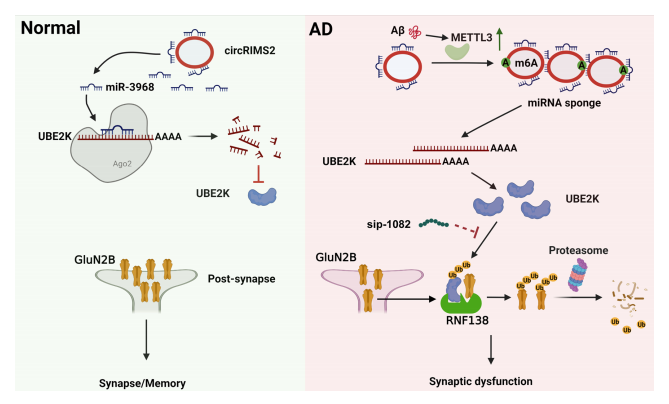
在本研究中,我们在AD中发现了一个异常上调的circRIMS2/miR-3968 ceRNA对。该通路的上调导致了UBE2K的增加和泛素化介导的GluN2B的降解,从而导致记忆和突触损伤。
此外,阻断circRIMS2/miR-3968/UBE2K/ GluN2B轴可显著改善APP/PS1小鼠的突触功能障碍。
总之,我们对AD早期阶段的circRNA/miRNA的表达模式进行了表征,其中小鼠有突触功能障碍,但没有明显的Aβ沉积。
我们揭示了上调的circRIMS2/miR-3968通路通过激活ube2k介导的GluN2B泛素化和降解,介导AD突触和记忆损伤的重要作用。
此外,我们还发现,阻断GluN2B的泛素化可以改善AD模型小鼠的突触功能障碍。我们的工作提出了几个新的治疗靶点来克服突触和记忆障碍。



作者使用伯信生物明星产品circRNA 表达载体、RAP、FISH以及探针进行了上述筛选与分子互作调控机制的研究。

