


(IF=15.1004)《npj | digital medicine》
摘要
透明细胞肾细胞癌(ccRCC)是一种侵袭性恶性肿瘤,与 VHL 、PBRM1和SETD2基因的单核苷酸变异相关。然而,结构变异(SV)——其具有更广泛的基因组影响——以及三维基因组结构在ccRCC中的作用仍不明确。
本文报道了该肿瘤的全面分子特征分析。通过多组学分析,我们鉴定出新型结构变异(SV)相关致癌靶点,并揭示了肾透明细胞癌(ccRCC)进展过程中多维三维基因组重组。
我们阐明了SV与三维染色质结构之间的动态相互作用,证明结构重排如何通过三维基因组重组驱动致癌失调。值得注意的是,我们发现并实验验证了一种未被识别的致病性增强子劫持事件,该事件导致原癌基因SEMA5B的组成型激活。
此外,我们开发了一个基于机器学习的预后框架,采用增强子劫持特征。总体而言,这项工作通过阐明结构变异(SVs)和三维基因组重组如何共同驱动肿瘤发生,为肾细胞癌(ccRCC)研究建立了宝贵的资源,并将这些发现转化为具有临床应用价值的预后工具。
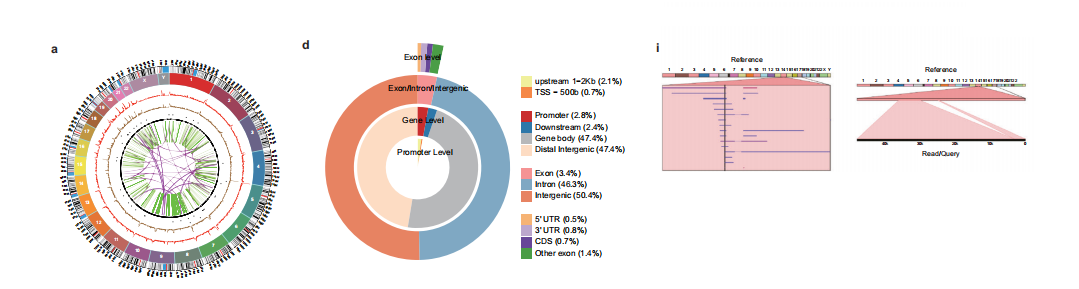
本研究采用纳米孔长读长全基因组测序技术,系统解析了与正常肾上皮细胞肿瘤发生相关的结构变异谱。
通过将测序数据与GRCh38(hg38)参考基因组比对,我们鉴定出大量高置信度的结构变异(SV)事件,并构建了详细的SV图谱。
在786-O、OS-RC-2和HEK293T细胞中,SV总计数分别为18,912、18,792和21,571。ccRCC表现出显著较低的重复率,这表明重复可能并非肾癌的决定性特征。
我们对不同结构变异(SV)类型长度分布的分析显示,插入和缺失事件呈现出相似的分布模式,峰值集中在300 bp附近,且大多数事件发生在1 kb范围内。
此外,对各染色体上不同SV类型标准化计数的检测表明,各类特定SV类型的分布模式基本一致。
我们随后研究了由重叠断点连接定义的复杂结构变异(SV)的流行情况。比较分析显示,提示癌变过程中基因组不稳定性增强。为鉴定可能驱动肿瘤发生的肿瘤特异性SV,我们对恶性与正常细胞系进行了系统性比较。
值得注意的是,约三分之一的SV表现出细胞系特异性,这表明谱系限制性SV在肾细胞癌(ccRCC)发病机制中具有功能相关性。
具有特殊临床意义的是,两种ccRCC模型均表现出HIF1A基因改变——该基因是低氧诱导型癌基因,受 VHL(肾癌守门基因)调控,其失调与不良预后相关27-29。
在786-O细胞系中,我们发现了一个38kb的基因内缺失,这与既往细胞遗传学和 NGS 研究报道的HIF1A缺失结果一致。相反,OS-RC-2细胞系则出现了一种新型重复事件,拓展了ccRCC中HIF1A改变的谱系。
为独立验证这些发现,我们使用整合基因组学浏览器(IGV)和Ribbon对所有三种细胞系的相关基因组区域进行了多平台可视化分析。
我们系统比较了第三代测序与新一代测序在检测ccRCC(肾透明细胞癌)结构变异(SVs)中的性能。长读长测序显示出更高的敏感性,识别出的高置信度SVs数量显著多于新一代测序。
全基因组ideogram分析显示断点密度显著增加。通过长读长测序技术检测到,尤其在着丝粒和端粒等复杂基因组区域。
重复序列相关缺失的注释分析表明,与新一代测序技术相比, TGS 检测到由重复元件驱动的结构变异(SVs)负担更高,且具有显著的组成特征:短散在核元件(SINEs)占 NGS 相关重复序列的58.8%,但在长读长数据中比例降低;而其他重复序列类型(如长散在核元件LINEs、长末端重复序列LTRs)则呈现更高占比。
TGS 识别的重复序列表现出更广泛的染色体分布和更大的平均长度。亚家族水平分类及多重复序列重叠的SV分析进一步验证了这些发现。
综上所述,本文鉴定的结构变异(SVs)直接影响癌基因和肿瘤抑制基因,可能对肿瘤恶性程度及肿瘤表型维持产生显著影响。
我们的长读长测序分析揭示了人类肾癌中SVs的分布图谱——这一先前研究不足的领域,并为未来关于肾细胞癌(ccRCC)发病机制的研究提供了宝贵资源。
为探究SVs对高阶染色质结构的全局影响,我们采用HiC技术对ccRCC细胞与正常细胞的三维基因组进行了分析。多尺度比较分析显示,恶性细胞的三维基因组结构相较于正常细胞存在显著重组。
染色体水平分析显示区室身份保守性趋势,稳定A区室在ccRCC细胞中占比最高。
与已知区室在基因调控中的作用一致,区室转换与基因表达变化显著相关:稳定A区室和B转A区室表现出更高表达水平,而A转B区室则呈现低转录水平,提示基因沉默。
进一步分析表明,B区向A区的区室转换虽然总体涉及的差异表达基因较少,但在转录激活的癌基因方面却显著富集。
这一现象在786-O和OS-RC-2细胞系中均具有统计学显著性,其中宇宙注释的癌基因BTG1作为典型代表。
此外,皮尔逊相关性热图显示,癌细胞系不仅存在广泛的区室重塑,还显著增强了A/B区室模式。
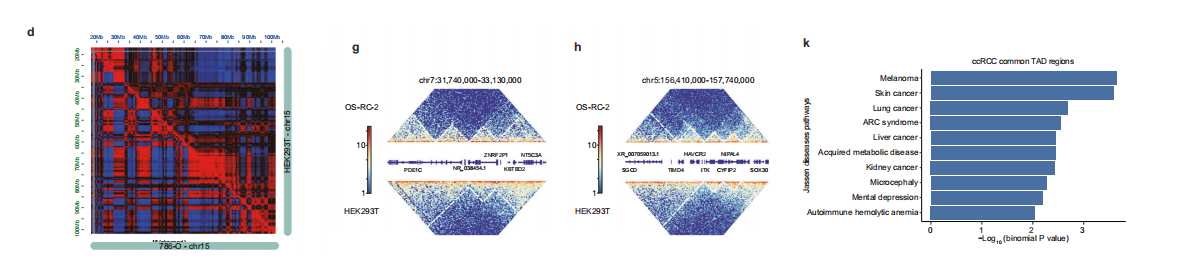
在拓扑关联结构域(TADs)的更精细尺度上,我们分别在786-O、OS-RC-2和HEK293T细胞系中以10 kb分辨率鉴定出7014、7823和7580个TADs。
值得注意的是,有3,907个TADs在所有三种细胞系中均保持保守,这种高度保守性凸显了人类基因组中TADs的稳定性,与既往研究15,32的结果一致。
为表征拓扑关联结构域(TAD)的改变,我们定义了ccRCC特异性TAD(肿瘤细胞中获得的)和ccRCC共用TAD(肿瘤细胞间共享但正常细胞中缺失的)。
结果显示,与保守TAD相比,ccRCC特异性及ccRCC共用TAD的尺寸显著减小,这一发现与既往关于多发性骨髓瘤、肝癌和前列腺癌等实体瘤的研究结果相似。
我们通过hicPlotTADs可视化交互矩阵来展示这些特征,结果清晰显示ccRCC细胞系中7号和5号染色体的TAD区域明显较小。
从功能上看,ccRCC特异性TAD中的基因表达水平显著高于保守TAD中的基因。此外,这些基因在肾脏癌及相关疾病通路中呈现显著富集,表明这些区域是转录失调和癌基因激活的热点区域。
综上所述,我们的研究结果表明,肾透明细胞癌(ccRCC)在区室和拓扑关联结构域(TAD)水平上均会发生广泛的癌症特异性三维基因组重塑。
这些结构变化与转录失调及癌基因的异常激活密切相关,提示其可能是ccRCC发生发展和进展的潜在驱动因素。
既往研究已证实,体细胞结构变异(SV)的形成受三维基因组结构影响并与其相互调节。为阐明这种关系在肾细胞癌(ccRCC)中的表现,我们系统研究了SV在不同层级染色质结构中的分布模式。
通过置换检验进行的初始区室水平分析显示,ccRCC特异性SV和细胞类型特异性SV在转录活跃的A区室中均呈现显著富集。
在局部基因组尺度上,癌细胞系(786-O和OS-RC-2)在A区室中表现出更高的缺失和插入密度,同时B区室中密度降低,这种模式在正常细胞中未观察到。
这些发现共同表明,结构变异(SV)在A区室中呈现优先积累现象,这可能与A区室高转录活性及由此导致的高双链断裂(DSB)发生率相关,同时也与B区室向A区室的动态转换机制存在关联。
肿瘤特异性结构变异(SV)分析显示,其分布模式与亲代细胞系相似,其中缺失/插入变异呈现A区室富集特征。值得注意的是,786-O特异性缺失在B区室中显著减少。
区室转换分析表明,该变异具有肾细胞癌(ccRCC)特异性。在B到A区域中持续富集的缺失事件,而插入和TAD水平的研究表明,边界富集的重复分布表现出细胞系特异性差异。
此外,跨区室类型的边界区域中,SV类型组成保持可比性,除786-O重复外,大多数SV类型的密度均有所升高。
OS-RC-2插入与优先级一致。肿瘤特异性结构变异(SVs)与TAD重塑存在细胞类型依赖性关联,其中OS-RC-2插入富集于结构破坏的边界区域。
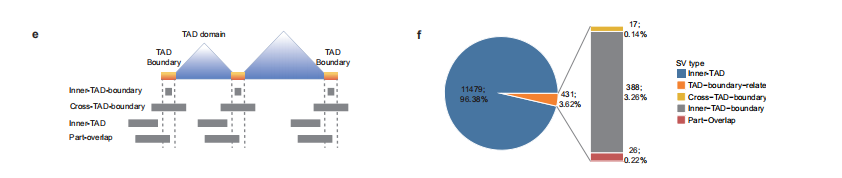
通过TAD定位对ccRCC特异性结构变异(SV)进行分类,共鉴定出四种不同类别。其中大多数变异位于TAD内部区域(内TAD),表明其对染色质折叠的影响较小。
具有破坏染色质折叠潜力的SV(即跨越TAD边界或部分重叠的变异)表现出独特的SV类型组成,其中跨越边界的事件以缺失为主。
这些结果共同表明,肾细胞癌中的结构变异分布与三维染色质结构存在非随机关联。我们的分析揭示了结构变异在转录活跃的A区室及其过渡区的优先积累,同时伴随TAD边界关联的细胞类型特异性偏好。
此外,结构变异对拓扑组织的破坏潜力似乎取决于变异类型,其中缺失主要影响跨越边界的区域。
体细胞结构变异与染色质结构之间观察到的空间协调性,提示在肾癌发生过程中基因组不稳定性与三维基因组组织之间存在复杂的相互作用。
空间基因组组织是染色体重排和肿瘤发生的关键决定因素。既往研究表明,结构变异(SVs)可通过破坏拓扑关联结构域(TAD)边界,诱导染色质结构改变。
此类扰动可能导致相邻TAD融合或活性染色质异常扩散,从而建立致癌性增强子-启动子相互作用。
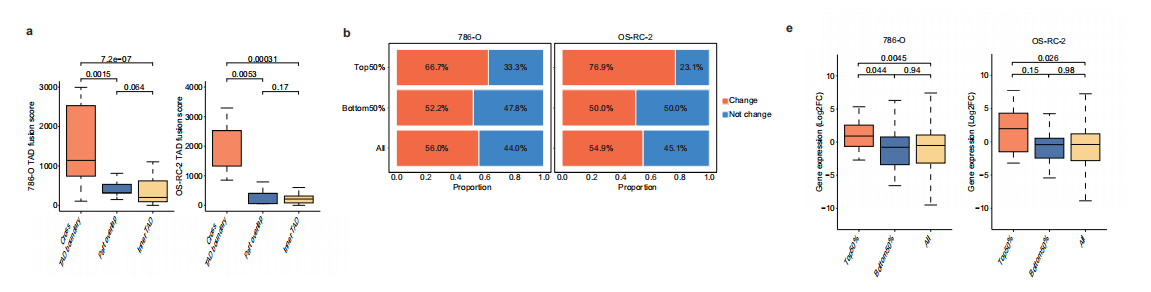
为探究SVs对肾细胞癌中TAD破坏的影响,我们分析了不同类别TAD缺失与TAD融合事件之间的相关性。
我们的研究结果表明,跨越TAD边界区域的缺失与其它类型缺失相比,表现出显著更高的TAD融合评分。
这些发现与先前研究结果一致,即跨越边界的缺失与TAD融合密切相关。值得注意的是,位于TAD内部区域的结构变异(SVs)——即便是体积较大的变异——对相邻染色质相互作用的破坏程度极小,邻近TAD基本不受影响。
相反,跨越TAD边界的SVs会显著增加缺失位点附近相邻TAD之间的染色质相互作用。我们进一步探究了两个癌细胞系中拓扑关联结构域(TAD)融合与转录失调之间的关系。
差异基因表达分析表明,与融合评分较低的区域或全基因组基线相比,携带TAD融合评分前50%结构变异(SV)的基因组区域含有显著更高比例的失调基因。
值得注意的是,高TAD融合评分区域内的基因表达水平较全基因组平均水平显著升高。这些观察结果表明,TADs维持基因组结构完整性,而其边界作为易损枢纽,易受结构变异驱动的致癌回路重塑影响。
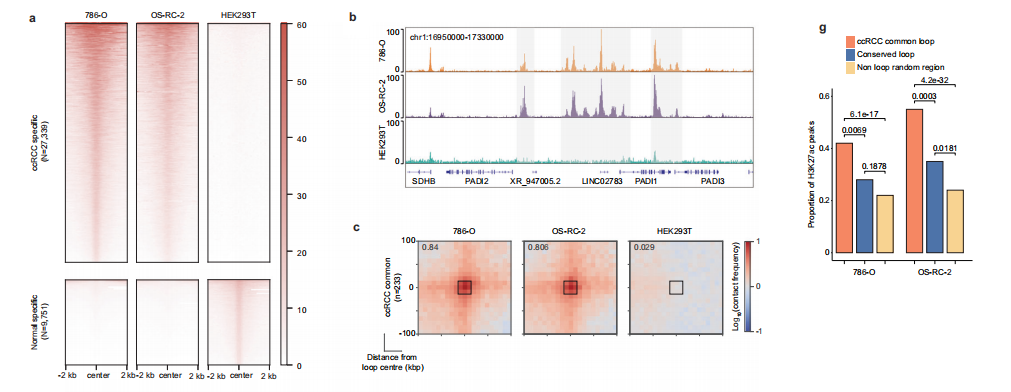
在完成大规模三维基因组结构分析后,我们进一步聚焦增强子景观与焦点染色质相互作用,以揭示ccRCC发病机制中的精细调控逻辑。
染色质状态比较分析证实了不同的增强子活性模式:ccRCC特异性增强子在肿瘤细胞系(786-O、OS-RC-2)中活跃,但在HEK293T细胞中不活跃;而正常组织特异性增强子则呈现相反模式。
代表性基因组位点的 IGV 图进一步验证了我们差异增强子识别方法的稳健性。通过GO数据集进行的肿瘤特异性增强子功能注释分析显示,这些增强子显著富集于ccRCC生物学核心通路,包括血管生成3和缺氧反应。
相比之下,正常特异性增强子则与泌尿系统发育过程相关,如中肾小管形成和输尿管芽形态发生。我们采用5 kb分辨率的Mustache技术对全基因组染色质互作进行分析。
差异分析显示,223个环状结构为ccRCC共性特征,55个为保守特征,377个为正常组织特异性特征。聚类峰分析表明,与正常组织相比,肿瘤样本中ccRCC共性互作信号呈现显著聚集。
值得注意的是,ccRCC共性环状结构的活性水平显著更高。与保守环和随机对照区相比,增强子信号更为显著。
染色质互作热图显示了ccRCC共有的长程互作特征,例如涉及MAP4K4、RFX8和CREG2的相互作用,这些区域均观察到肿瘤共有的增强子信号。
为深入探究这些重构互作的影响,我们将染色质互作数据与转录组谱进行整合分析。结果显示,与所有已识别环或保守环相比,ccRCC共用环的锚点区域与显著更高的基因表达水平相关。
综上所述,我们的研究结果表明,ccRCC的发病机制涉及广泛的增强子重编程及局部染色质相互作用的重塑。
在ccRCC特异性环区中活性增强子的富集及其与基因表达升高的强关联性,突显了精细尺度三维基因组重构在驱动肾癌发生过程中转录失调的作用。
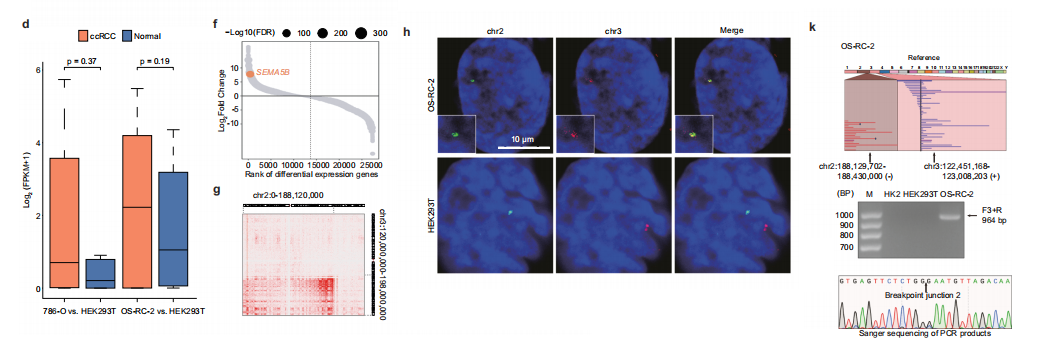
最新研究发现,结构变异(SVs)可通过增强子劫持机制异常激活癌基因——该机制使SVs将增强子元件重新定位至致癌启动子附近。
为系统识别肾细胞癌(ccRCC)中的此类事件,我们应用了Neoloopfinder工具。
基于 TGSWGS 和Hi-C校正的基因组拷贝数变异(CNVs)提供的全面共识SV数据,我们在SV断裂点附近鉴定出激活癌基因的异位染色质相互作用,称为新环(neoloops)。
聚类峰分析显示,与正常组织对照相比,ccRCC中新环信号存在显著富集。转录组分析显示,肿瘤细胞系中位于新环锚定区域的基因表达水平显著升高,这证实了这些异常染色质相互作用在致癌基因激活中的功能相关性。
在已识别的新环结构中,我们发现了一个复杂的结构变异(SV):通过协调的染色体倒位和易位,使远端增强子簇(chr3:128,620,000-128,930,000)相对于SEMA5B启动子(chr3:122,450,000-123,000,000)发生近端重定位。
Hi-C接触图谱证实,该位点在正常细胞中完全不存在启动子-增强子相互作用,而肿瘤样本则表现出与SEMA5B转录上调同步的显著染色质连接性。
为验证这些结构变异(SVs)的物理存在性,我们进行了实验验证。采用断点侧翼探针的双色DNA荧光原位杂交(DNA-FISH)显示,2号和3号染色体片段在肿瘤中呈现特异性共定位,证实了该易位的体细胞起源。
通过断点连接PCR及桑格测序,成功扩增并验证了肿瘤细胞系中的杂合性倒位/易位事件。单分子长读长测序数据进一步佐证了这些发现,显示仅在肿瘤样本中存在跨越两个重排连接点的连续DNA读长。
在所有实验模式中,匹配的正常细胞系均未检测到这些结构重排的证据。
综上所述,我们的研究结果表明,ccRCC中的结构变异(SVs)可通过破坏局部染色质结构来介导长距离增强子劫持,从而形成致癌性增强子-启动子环。
这种三维基因组重组通过异常结合增强子与新靶基因,促进了肿瘤特异性转录激活。此外,我们的多平台验证框架为这些致病性重连事件背后复杂的SVs的存在提供了确凿证据。
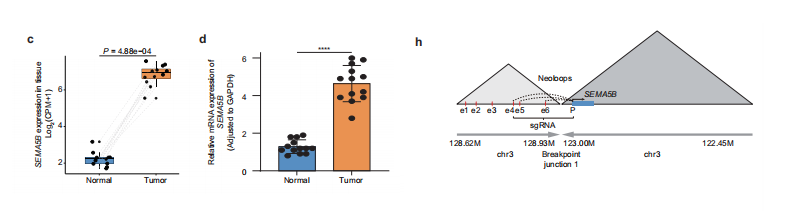
尽管信号素(semaforins)正逐渐成为癌症临床生物标志物和治疗靶点,但其在肾细胞癌(ccRCC)中的特异性致癌作用仍缺乏明确表征。
为填补这一研究空白,我们首先利用癌症基因组图谱(TCGA)评估了SEMA5B在泛癌数据集中的表达情况。
结果显示,SEMA5B在脑组织和肾组织中呈现高表达特征,且与正常肾组织相比,肾癌组织中其表达水平显著升高,提示SEMA5B在ccRCC中存在特异性过表达现象。
进一步分析显示,SEMA5B在HK-2和HEK293T等正常细胞系中转录组和蛋白表达水平较低,而OS-RC-2肾癌细胞中表达显著升高。
通过RNA-seq和RT-qPCR在临床样本中验证了这些发现,证实SEMA5B在转录组水平呈现肿瘤特异性过表达。
免疫组化(IHC)进一步证实SEMA5B在蛋白水平的肿瘤特异性上调。
为探究SEMA5B过表达的调控机制,我们基于neo-loop结构坐标鉴定了参与异位相互作用的增强子(e4–e6)。
针对这些增强子及SEMA5B启动子区域设计了靶向sgRNA,用于 CRISPR 干扰(CRISPRi)实验,并合成了靶向SEMA5B外显子的siRNA。
通过蛋白质印迹和RT-qPCR验证了敲低(KD)及CRISPRi的效率。靶向异位增强子的CRISPRi显著降低了SEMA5B表达,验证了neo-loop鉴定的可靠性及其转录调控效应。
我们进一步评估了破坏SEMA5B异位增强子和启动子的表型影响。体外实验显示,这些调控元件的缺失显著抑制了细胞增殖和侵袭能力,该结果与敲除(KD)实验数据一致。
体内实验进一步证实,SEMA5B异位增强子和启动子的破坏可显著减缓肿瘤增殖。
综上所述,我们通过整合临床队列、细胞模型和功能基因组学的分析,证实SEMA5B是一种由增强子劫持激活的肾透明细胞癌(ccRCC)特异性癌基因。
体外和体内模型的一致表型证据强调了这种新环介导的转录重编程在ccRCC发病机制中的关键作用,使SEMA5B成为肾恶性肿瘤的潜在治疗靶点。
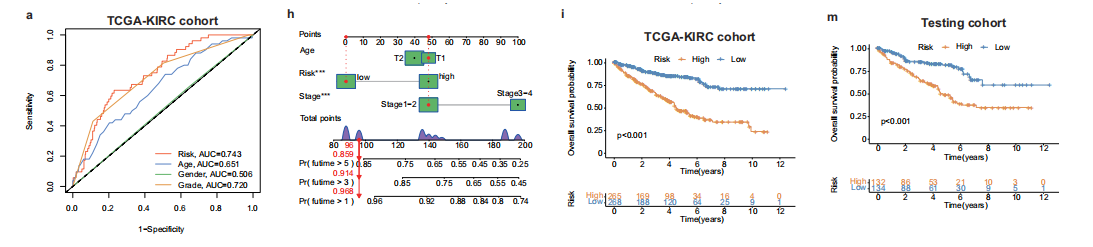
为评估研究结果的临床应用价值,我们基于ccRCC特异性新发肿瘤相关基因,利用 TCGA - KIRC 队列开发了机器学习模型。
该模型展现出优异的预测性能:训练队列曲线下面积(AUC)达0.747,测试队列0.740,完整患者队列0.743,均显著优于传统WHO分级系统(AUC值分别为0.745、0.703和0.720。
此外,该模型运行稳定且模型在1至5年随访期间保持持续的预测准确性。我们还构建了一个整合多项临床参数的列线图,用于预测ccRCC患者的总生存期(OS)。校准曲线分析证实了该模型的稳健预测性能。
基于模型推导的风险评分,患者被划分为明确的高风险组和低风险组,两组生存结局存在显著差异。
Kaplan-Meier法分析显示,无论是训练队列还是完整 TCGA - KIRC 队列,高风险组与低风险组的总生存期(OS)和无进展生存期(PFS)生存曲线均呈现显著分异,这表明模型能有效区分不同风险水平的患者。
值得注意的是,这种显著的生存曲线分异在测试队列中也得到一致验证,证实了该模型在不同患者亚组中均具有稳健性和预测准确性。
值得注意的是,模型推导的风险评分与既定的疾病严重程度指标呈显著正相关。疾病进展更严重的患者——包括年龄较大、病理 TNM 分期较高、临床分期较高及WHO分级较高者——其风险评分均持续升高。
这些关联进一步验证了我们基于neoloop的预后模型的临床相关性和生物学一致性。
综上所述,我们的多组学方法揭示了结构变异(SVs)如何重塑三维染色质组织以激活肾细胞癌(ccRCC)中的致癌通路,从而弥合了结构基因组改变与转录失调之间的鸿沟。
尽管本研究提供了关键性见解,样本量限制及缺乏纵向数据等局限性仍需进一步研究。未来工作应探索肿瘤演变过程中结构变异(SV)积累的时间动态特征,并评估靶向增强子劫持事件的治疗效果。
通过阐明SV与三维基因组组织之间的相互作用,本研究不仅深化了对肾透明细胞癌(ccRCC)生物学机制的理解,也为精准肿瘤学开辟了新途径。
由此建立的风险预测模型将这些机制性见解转化为临床适用工具,有助于优化患者分层策略。

作者使用伯信生物明星产品DNA FISH试剂盒和Probes进行分子调控机制研究。
伯信好物推荐
DNA FISH Kit
伯信生物建立了荧光原位杂交探针(FISH Probe)制备平台,承接荧光原位杂交试剂盒定制业务,可根据客户需求进行个性化定制。
该试剂盒主要用于检测样本中目的基因的表达量与表达部位,可应用于组织切片(石蜡切片/冰冻切片)、细胞爬片与染色体片等的检测。伯信生物采用从研究物种自体获取特异性探针并进行化学共价方式与荧光素连接。
针对目的DNA片段设计特异性探针并进行DNA-FISH,可应用于DNA染色体定位、基因拷贝数、染色体结构、转基因分析等。

伯信DNA FISH Kit分为:
Bes1011(S) DNA FISH Kit 30T
Bes1011 (N) DNA FISH Kit 50T
Bes1011 (M) DNA FISH Kit 100T
实验原理:
荧光原位杂交是根据待测核酸序列设计特异性的荧光素标记寡聚核苷酸探针,再经过共变性-退火-复性,使探针与靶DNA按照碱基互补配对原则形成杂交体,最后利用荧光显微镜直接检测荧光信号,从而对组织、细胞中的待测核酸进行定性、半定量或相对定位分析的一种原位杂交技术。
技术流程:


结果实例:

产品优势:
1. 探针制备采用直接标记法,省去了信号放大系统。
2. 荧光信号强,结果判定直观可靠。
3. 根据客户需求定制个性化探针。
4. 特异性强,灵敏度高,背景低。
5. 探针性能稳定,低温保存一年以上。
6. 同时示踪核酸和蛋白的定位情况。
7. 操作简单,安全、快速,重复性好。

