


Exosomal long non-coding RNA TRPM2-AS promotes angiogenesis in gallbladder cancer through interacting with PABPC1 to activate NOTCH1 signaling pathway
《Molecular Cancer》(IF=37.2999)
背景:异常血管生成对胆囊癌(GBC)肿瘤的生长和侵袭至关重要,强调了阐明这一过程的机制的重要性。
LncRNA(长链非编码RNA)广泛参与了GBC的恶性肿瘤。然而,目前还缺乏关于GBC中lncrna与血管生成相关性的确凿证据。
方法:采用LncRNA测序,鉴定不同表达的LncRNA。采用RT-qPCR、western blot、FISH和免疫荧光法检测TRPM2-AS和NOTCH1信号通路的体外表达。
采用小鼠异种移植和肺转移模型评价TRPM2-AS在体内血管生成过程中的生物学功能。采用EDU、transwell和试管形成法检测HUVECs的血管生成能力。
利用RIP、RAP、RNA下拉、双荧光素酶报告系统和质谱分析TRPM2-AS、IGF2BP2、NUMB和PABPC1之间的相互作用。
结果:TRPM2-AS在GBC组织中表达上调,并与GBC患者的血管生成和预后不良密切相关。TRPM2-AS的高表达水平和稳定性得益于m6 A的修饰,这已被IGF2BP2识别。
在发挥促血管生成作用方面,从GBC细胞转运的外泌体TRPM2-AS增强了pabpc1介导的NUMB表达抑制,最终促进NOTCH1信号通路的激活。
PABPC1通过与AGO2相互作用,抑制NUMB mRNA的表达,促进miR-31-5p和miR-146a-5p介导的NUMB mRNA的降解。
NOTCH信号通路抑制剂DAPT抑制了GBC肿瘤血管生成,而TRPM2-AS的下调增强了这一效果。
结论:TRPM2-AS是一种新颖的、很有前途的GBC血管生成生物标志物,通过促进NOTCH1信号通路的激活来促进血管生成。靶向TRPM2-AS为未来的GBC治疗提供了更多的机会。

为了探讨GBC中血管生成的机制,我对血管密度最高和最低的3个GBC组织进行lncRNA测序。
RT-qPCR检测了12对低密度血管GBC组织和高密度血管GBC组织中P值最明显的前10个lncrna。
结果显示,在高微血管密度的GBC组织中,只有TRPM2-AS显著上调。
进一步的RT-qPCR和FISH实验显示TRPM2-AS表达上调与匹配的正常组织相比,高微血管密度的GBC组织中TRPM2-AS明显高于低微血管密度的GBC组织。
线性回归分析也表明,TRPM2-AS的表达与微血管密度呈正相关。
Kaplan-Meier分析显示,TRPM2-AS与GBC患者的总生存期(OS)和无复发生存期(RFS)降低之间存在相关性。
通过单因素和多因素Cox回归分析,我们发现TRPM2-AS表达水平是GBC患者的独立预后因素。
因此,这些结果提示TRPM2-AS在GBC组织中高表达,并与GBC组织的微血管密度和GBC患者的不良预后密切相关。
此外,我们通过核分离和FISH分析确定了TRPM2-AS的亚细胞定位,结果表明TRPM2-AS主要定位于GBC细胞的细胞质。
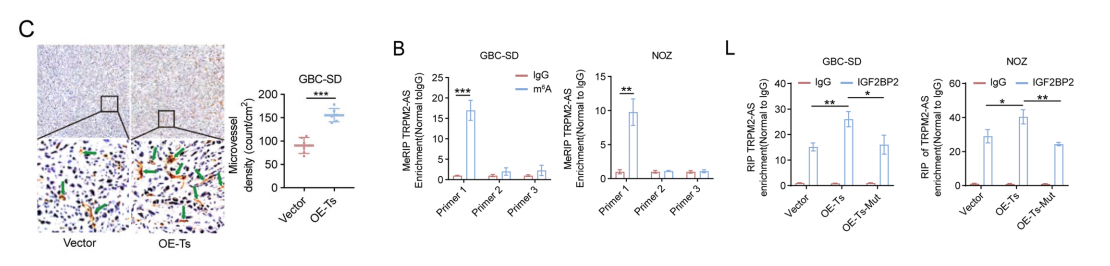
我们想知道TRPM2-AS在GBC上是否有促血管生成的作用。
异位接种过表达TRPM2-AS的GBC细胞,肿瘤生长速度更快,显著增加终点肿瘤重量和体积,以及切除肿瘤的微血管密度。
此外,我们在肺转移模型中获得了类似的结果。
TRPM2-AS过表达组图2E肿瘤中荧光素酶活性、肺转移结节数量和结节微血管密度均显著增加,而TRPM2-AS下调组消除了荧光素酶活性、肺转移、结节血管密度增加的优势。
m6 A甲基化是最常见的RNA修饰,我们使用SRAMP预测了TRPM2-AS中m6 A甲基化位点的存在,并确定了三个可能的m6 A甲基化位点。
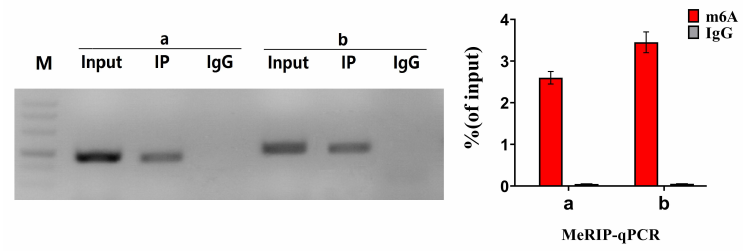
MeRIP检测结果显示,TRPM2-AS组中有271个位点发生了m6 A甲基化。
我们在GBC细胞中过表达了M3/M14,发现两种m6 A均发生了甲基化和TRPM2-AS的表达水平显著升高。
以上实验表明,增加m6 A对TRPM2-AS的修饰可以增加TRPM2-AS的表达。m6 A对RNA甲基化的影响在很大程度上依赖于m6 A阅读器。
为了鉴定介导TRPM2-ASm6A修饰的m6 A阅读器,我们进行了RIP检测,在这些读者中,TRPM2-AS仅在IGF2BP2组中高度富集。
双荧光素酶报告基因分析显示,IGF2BP2直接与TRPM2-AS相互作用。我们构建了IGF2BP2过表达和敲除GBC细胞系。
我们使用actinomycin D抑制整体RNA合成,果显示,过表达IGF2BP2后,TRPM2-AS的稳定性和表达水平升高,而下调IGF2BP2后,其稳定性和表达水平显著降低。
因此,IGF2BP2与TRPM2-AS相互作用,促进TRPM2-AS的稳定性。
为了验证m6 A的修饰是IGF2BP2和TRPM2-AS相互作用的基础,我们用DAA处理GBC细胞,以抑制TRPM2-AS的m6 A甲基化。
RIP检测结果显示,IGF2BP2富集的TRPM2-AS水平明显降低,此外,我们合成了m6 A修饰的RNA探针并进行了S1m下拉实验,当271位点的核苷酸发生突变时,IGF2BP2蛋白水平显著下降。
因此,m6 A修饰位点的271位点对于IGF2BP2与TRPM2- AS的结合是必不可少的。接下来,我们转染了TRPM2-AS和突变的TRPM2-AS过表达质粒。
RIP结果显示,过表达TRPM2-AS也显著提高了igf2bp2结合的TRPM2-AS水平。
这些结果强烈表明,IGF2BP2通过介导TRPM2-AS的m6 A修饰,增加了TRPM2-AS的稳定性。
与TRPM2-AS在GBC细胞中的致癌和促血管生成作用相一致,单因素和多因素Cox回归分析显示,IGF2BP2是患者预后的独立预后因素。
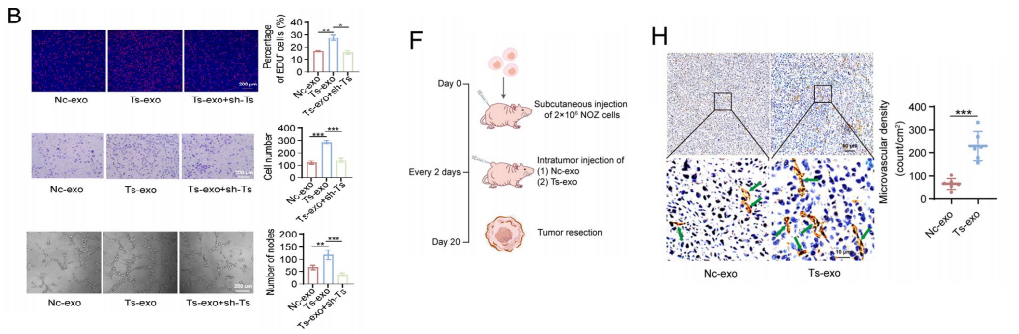
为了研究外泌体是否介导了TRPM2-AS从GBC细胞转移到HUVECs,我们选择了由NOZ细胞产生的外泌体进行实验。
我们通过PKH26标记外泌体并与HUVECs共培养,研究了TRPM2- AS从GBC细胞到HUVECs的运输。
结果显示,外泌体将TRPM2- AS转运到HUVECs.EDU的结果,transwell和管形成分析显示,增殖,迁移,和管形成能力HUVECsTs-CM和Ts-exo组明显高于Nc-CM组。
我们还观察到BCL-2、cyclinD1和VEGFA在Ts-CM和Ts-exo组中的表达增加,而BAX的表达显著降低。
因此,外泌体是TRPM2-AS促进血管生成的关键介质。
我们将从对照NOZ细胞和Ts-exo中提取的对照外泌体与HUVECs共培养,并使用shRNA逆转Ts-exo诱导的TRPM2-AS过表达。
与Nc-exo组相比,Ts-exo组的HUVECs的增殖、迁移和血管生成显著增强,而TRPM2-AS敲低逆转了这一缺陷。
我们发现32例患者血清中提取的外泌体中TRPM2-AS水平与肿瘤组织的微血管密度呈正相关。
因此,Ts-exo通过传递TRPM2-AS而不是其他蛋白质或RNA分子来促进GBC细胞中的血管生成。
为了检测外泌体TRPM2-AS是否促进体外血管生成,构建异种移植瘤模型。Tsexo治疗导致皮下肿瘤生长速度更快,终点肿瘤更重、更大。
在微血管密度方面,CD34免疫组化显示Ts-exo组的微血管密度明显高于Nc-exo组。
肺转移模型显示Ts-exo治疗导致了肺转移的显著差异,表现为更多的肺转移。
CD34免疫组化也显示,Ts-exo组转移性肿瘤的微血管密度明显高于Nc-exo组。
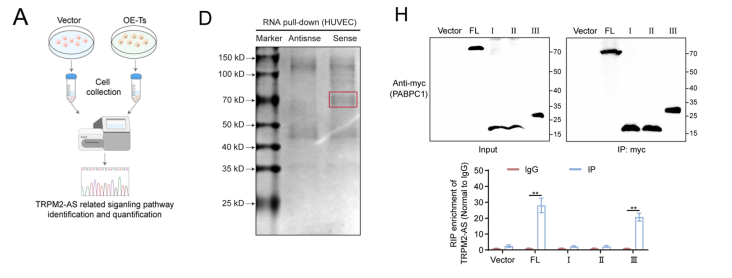
我们试图探索TRPM2-AS促进HUVECs增殖、迁移和试管形成的机制。因此,我们进行了转录组测序。
在TRPM2-AS相互作用的预测信号通路中,NOTCH信号通路被明显激活。过表达TRPM2-AS显著增加了NOTCH1信号通路中关键分子的表达水平。
与Nc-exo组相比,Ts-exo组的NOTCH1信号通路被激活,而TRPM2-AS敲低则显示了相反的结果。
我们使用DAPTNOTCH1信号通路的阻断剂,抑制γ分泌酶的功能,从而抑制NOTCH1信号通路的激活。
DAPT处理显著逆转了TRPM2-AS过表达和Ts-exo诱导的NOTCH1信号通路的激活。
DAPT显著抑制了TRPM2-AS过表达和Ts-exo诱导的HUVECs增殖、迁移和血管生成的促进作用。
与GBC细胞一致,FISH和核质分离显示,在HUVECs中,TRPM2-AS主要分布在细胞质中。
因为lncRNA通过海绵化miRNA或与蛋白结合来发挥作用,我们进行了RNA下拉分析,并结合质谱,以确定直接与TRPM2-AS相互作用的蛋白质。
与反义组相比,在70 kD位置的蛋白表达水平有显著差异。进一步检测HUVECs中NOTCH1信号通路的激活情况。
在这些蛋白中,只有PABPC1敲低抑制了NOTCH1信号通路,而过表达PABPC1激活了NOTCH1信号通路。
过表达PABPC1显著促进了HUVECs的增殖、迁移和管状形成。这些结果表明,PABPC1是NOTCH1激活的关键分子TRPM2-AS诱导HUVECs增殖、迁移和管形成。
我们根据二级结构合成全长生物素标记的TRPM2-AS及其片段探针。RNA下拉分析显示,只有当使用全长和片段C作为探针时,才能检测到PABPC1。
为了进一步鉴定关键结构域,我们构建了过表达myc标记的PABPC1全长和片段I、II和III的质粒。
RNA下拉结果显示,只有TABPC1全长和片段III被TRPM2-AS拉下。我们使用抗myc抗体进行了RIP实验。
结果显示,TRPM2-AS在PABPC1全长组和片段III组中高度富集。综上所述,TRPM2-AS通过结构域C与PABPC1的片段III相互作用。

我们发现TRPM2-AS与NUMB mRNA的3‘UTR之间存在一个互补序列,这是NOTCH1信号通路的典型抑制因子。
TRPM2-AS和PABPC1的过表达导致NUMB mRNA和蛋白的表达水平降低。
我们应用RAP检测发现用蛋白酶K从底物中去除蛋白,显著降低了TRPM2-AS探针拉下的NUMB mRNA水平。
因此,TRPM2-AS和NUMB mRNA之间的相互作用依赖于蛋白质配对,而不是碱基互补配对。RIP检测证明NUMB mRNA与PABPC1直接相互作用。
值得注意的是,PABPC1是TRPM2-AS和NUMB mRNA之间的桥梁。
我们使用生物素标记的TRPM2-AS探针进行RAP实验,结果显示,在对照组中,过表达TRPM2-AS的水平显著提高了下拉产物中TRPM2-AS和NUMB的水平。
western blot也证实了PAPBC1是TRPM2-AS与NUMB之间的中介分子:下调PABPC1后,TRPM2-AS对NUMB蛋白表达的抑制作用消失。
因此,PABPC1对于TRPM2-AS与NUMB mRNA的相互作用是必不可少的。
TRPM2-AS是否影响PABPC1和NUMB mRNA之间的相互作用尚不清楚。
在HUVECs中使用抗PABPC1抗体的RIP检测显示,在HUVECs中,TRPM2-AS下调后,PABPC1下调的NUMB mRNA水平显著降低。
TRPM2-AS的敲除进一步削弱了pabpc1介导的对NUMB表达的抑制,NOTCH1信号通路的激活和HUVECs血管生成。
综上所述,TRPM2-AS增强了PABPC1与NUMB mRNA区域的相互作用,从而增强了PABPC1对NUMB mRNA的抑制,最终激活NOTCH1信号通路,促进GBC中的血管生成。
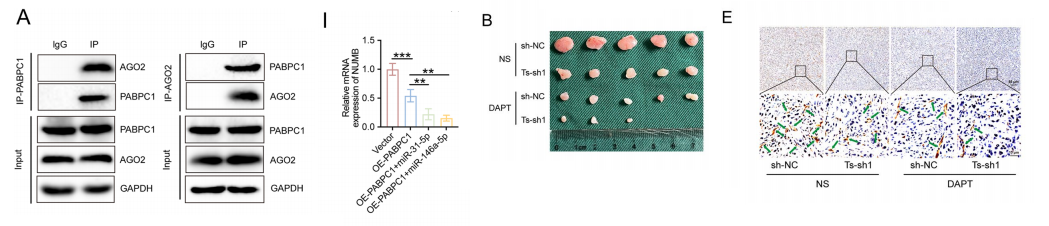
由于PABPC1抑制了HUVECs中NUMB的表达,我们推测mirna介导的mRNA降解可能参与了这一过程。
共免疫共沉淀分析显示,在HUVECs中,PABPC1与AGO2直接相互作用。双荧光素酶报告基因检测结果显示,miR-31-5p和miR-146a-5p与NUMB mRNA结合。
转染miR-31-5p和miR-146a-5p模拟物后,NUMB的mRNA和蛋白水平均被下调。PABPC1过表达显著促进了miR- 31-5p和miR-146a-5p的抑制作用,而PABPC1则降低了抑制作用。
此外,我们在PABPC1过表达的基础上增加了miR-31-5p和miR-146a-5p的表达,miR-31-5p和miR-146a-5p改善了PABPC1的这些功能。
此外,当敲除AGO2以限制mirna介导的mRNA降解时,pabpc1介导的NUMB表达抑制、NOTCH1信号通路激活和HUVECs血管生成均受到显著限制。
实验结果表明,PABPC1通过mirna介导的NUMB降解来抑制NUMB的表达。
我们推测TRPM2-AS可能对DAPT对GBC血管生成的抑制作用有协同作用。
建立小鼠异种移植和肺转移模型,TRPM2-AS敲低或DAPT单独限制了切除后肿瘤的生长,并降低了微血管密度肿瘤中,两者联合治疗导致肿瘤重量和体积的终点以及微血管密度有更明显的降低。
对于裸鼠肺转移,与单独敲除组和单独治疗组相比,H&E表明,两种联合治疗减少了肺组织中的转移性结节。
此外,免疫组化染色显示,TRPM2- AS和DAPT联合治疗组的微血管密度最低。TRPM2-AS的下调显著改善了NOTCH1阻断剂DAPT对异种移植瘤和肺转移模型中GBC血管生成的抑制作用。
总之,我们阐明了TRPM2-AS在GBC血管生成中的关键作用,并证明了IGF2BP2-TRPM2-AS/ PABPC1-NUMB-NOTCH1在GBC中是一个新的促血管生成轴。
TRPM2-AS过表达与GBC患者的血管生成和不良预后呈正相关。IGF2BP2通过m6 A依赖的机制确保了TRPM2-AS的稳定性和高表达。
值得注意的是,TRPM2-AS从GBC细胞运输到HUVECs,在那里它增强了PABPC1对NUMB mRNA表达的抑制作用,促进NOTCH1信号通路的激活,最终诱导HUVECs的增殖和迁移。
PABPC1通过与AGO2相互作用抑制NUMB mRNA的表达,促进miR-31-5p和miR-146a-5p介导的NUMB mRNA的降解(图10)。
靶向TRPM2-AS有望成为抑制GBC肿瘤血管生成的一种很有前途的策略,并可能为未来的GBC治疗提供更广泛的应用。




作者使用伯信生物明星产品RNA pulldown、RAP、RIP、meRIP试剂盒以及分子探针进行了上述筛选与分子互作调控机制的研究。
伯信好物推荐
RNA pulldown Kit
产品介绍:
1
蛋白质与RNA的相互作用是许多细胞生物学过程的核心,如蛋白质合成、mRNA 组装、病毒复制、细胞发育调控等,研究它们之间相互作用的分子机制对理解这些生物学过程非常重要。
RNA pulldown技术通过体外转录标记生物素的RNA探针耦联链霉亲和素标记的磁珠,实现了对RNA结合蛋白质(RBPs)的高效富集和鉴定。
伯信RNA pulldown Kit 分为:
Bes5102(S) RNA pulldown Kit 12T
Bes5102(N) RNA pulldown Kit 40T
实验原理:
针对目标区域设计生物素标记的特异性RNA探针偶联磁珠,并与细胞蛋白提取物孵育,蛋白分子与RNA探针特异性结合,经洗脱,得到目的RNA探针-蛋白质复合物。最后采用Western Blot或质谱鉴定蛋白质类型。
技术流程:
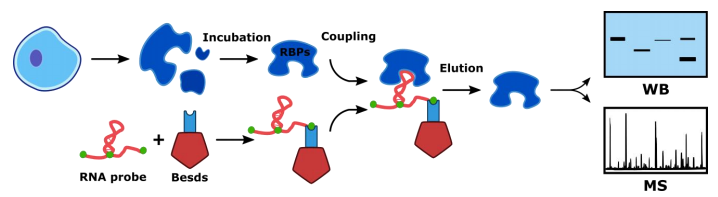
结果实例:

产品优势:
1. 自主品牌:拥有自主知识产权专利。
2. 高品质,稳定性好。
3. 精准:特异性高,背景少。
4. 易用:操作简单,容易上手。
5. 快速省时:全部实验只需3小时。
伯信好物推荐
RAP Kit
产品介绍:
2
RNA 反义纯化技术(RNA Antisense Purification,RAP)用于研究RNA与RNA、RNA与蛋白质之间的相互作用。根据研究对象不同,RAP技术结合了高通量测序(RAP-Seq)和质谱技术(RAP-MS),分别研究与目标RNA互作的RNA和蛋白质。
伯信RAP Kit 分为:
Bes5103-1(S) RAP-RNA Kit 12T
Bes5103-1(N) RAP-RNA Kit 30T
Bes5103-2(S) RAP-Protein Kit 12T
Bes5103-2(N) RAP-Protein Kit 30T
Bes5103-3(S) RAP-RNA、Protein Kit 12T
Bes5103-3(N) RAP-RNA、Protein Kit 30T
实验原理:
RAP技术通过设计生物素探针组拉取目标RNA,使与其共同作用的RNA或蛋白质(RBPs)富集在磁珠上并被洗脱。最后,通过高通量测序得到该调控RNA转录调控的下游靶基因,同时,也可以通过Western Blot验证RBPs,或通过MS鉴定未知蛋白。
技术流程:

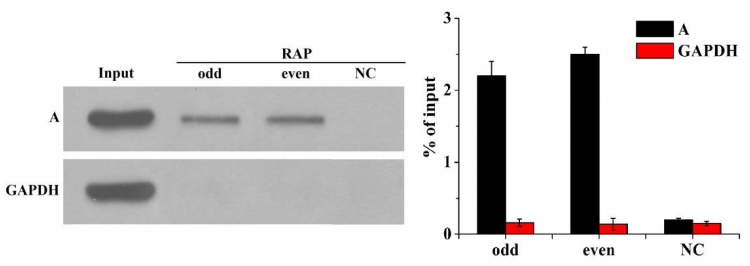
结果实例:
产品优势:
1. 伯信独立研发,具有自主知识产权。
2. 灵敏度高,稳定性好。
3. 检测方法灵敏,结果准确、重复性好。
伯信好物推荐
RIP Kit
产品介绍:
3
RNA Immunoprecipitation(RIP)是研究细胞内RNA与蛋白质结合的技术,是了解转录后调控网络动态过程的有力工具,可应用于miRNA调控靶点、RNA 与 RBPs(RNA结合蛋白)互作等研究。
伯信RIP Kit 分为:
Bes5101(S) RIP Kit 12T
Bes5101(N) RIP Kit 40T
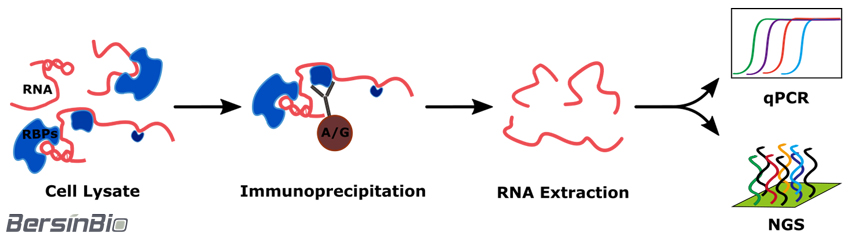
技术流程:
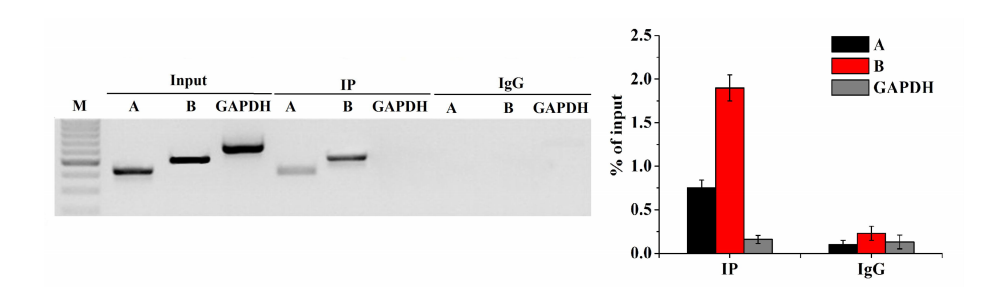
结果实例:
产品优势:
1. 伯信独立研发。
2. 提供优化的磁珠,适于免疫沉淀RNA-蛋白复合物。
3. 高品质,稳定性好,
4. 结果准确,重复性好。
5. 操作简单,快速省时。
伯信好物推荐
m6A MeRIP Kit
产品介绍:
4
RNA甲基化指发生在RNA分子上不同位置的甲基化修饰现象,属于转录后修饰,常见的甲基化修饰方式有6-甲基腺嘌呤(N6-methyladenosine,m6A)和5-甲基胞嘧啶(C5-methylcytidine,m5C)以及7-甲基鸟嘌呤(N7-methylguanosine,m7G)等。
RNA甲基化在调控基因表达、剪接、RNA编辑、RNA稳定性、控制mRNA寿命和降解、介导环状RNA翻译等方面可能扮演重要角色。
利用甲基化RNA免疫共沉淀(Methylated RNA Immunoprecipitation,MeRIP)技术,可以对RNA转录后甲基化修饰图谱进行全面研究。

伯信MeRIP Kit 分为:
Bes5203-1(S) MeRIP Kit 12T
Bes5203-1(N) MeRIP Kit 40T
Bes5203-2(S) MeRIP Kit(含m6A抗体) 12T
Bes5203-2(S) MeRIP Kit(含m6A抗体) 40T
实验原理:
MeRIP利用m6A特异性抗体识别并结合mRNA上的m6A甲基化修饰位点,以RNA免疫共沉淀方法富集甲基化修饰片段,同时,结合高通量测序与qPCR,在全转录组范围内研究发生m6A甲基化修饰的RNA区域,及其对基因表达的调控作用。
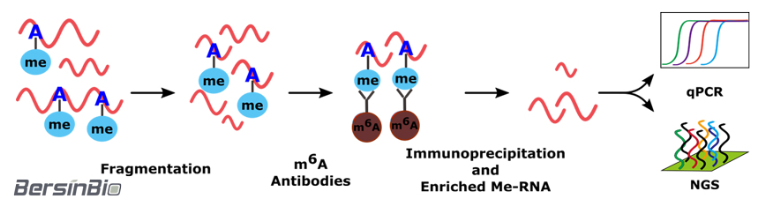
技术流程:
结果实例:
产品优势:
1. 快速省时
2. 稳定可靠
3. 配备完整

