


KAT2A-driven succinylation of SRSF11 enforces spliceosome mediated RAD52 splicing to promote homologous recombination and radioresistance in hepatocellular carcinoma
《Signal Transduction and Targeted Therapy》(IF=52.7006)
摘要
翻译后修饰中的琥珀酰化在多种恶性肿瘤的肿瘤发生过程中起着关键作用,但其在肝细胞癌(HCC)发病机制及治疗耐药性中的作用机制仍知之甚少。
本研究系统证实,剪接因子SRSF11在 HCC 进展过程中会发生功能性琥珀酰化修饰。从机制上看,赖氨酸乙酰转移酶2A(KAT2A)直接与SRSF11相互作用,催化其第419位赖氨酸(K419)的琥珀酰化,从而在体外和体内 HCC 模型中均增强了DNA损伤修复能力。
结构与功能分析表明,K419琥珀酰化修饰可稳定SRSF11与剪接体的相互作用,通过增强前体mRNA结合促进RAD52外显子10的包含。
这种外显子特异性剪接事件保留了同源重组(HR)修复所必需的RAD51结合域,最终促进RAD52-RAD51二聚体组装及HR介导的基因组稳定化。
临床上,SRSF11表达升高与HR活性增强、放射抗性增加及 HCC 患者生存率降低相关。值得注意的是,KAT2A-SRSF11轴的遗传学破坏会增强 HCC 细胞对辐射诱导凋亡的敏感性。
本研究发现琥珀酰化修饰是 HCC 中将可变剪接与DNA修复保真度联系起来的新型调控机制,同时提出通过靶向该通路治疗晚期 HCC 以克服放射抗性的策略。
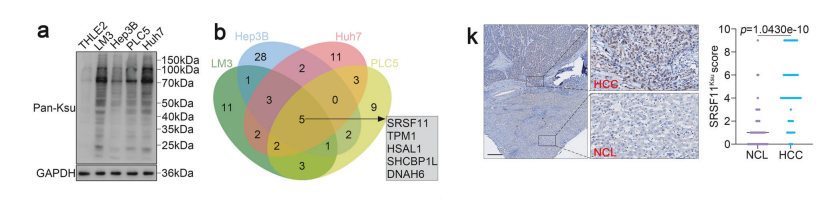
KAT2A驱动 HCC 中致癌性SRSF11蛋白K419位点的琥珀酰化修饰。全局琥珀酰化检测显示,与THLE2细胞相比, HCC 细胞系存在显著的过度琥珀酰化现象。
LC-MS/MS分析确定K419是SRSF11蛋白上主要的琥珀酰化位点。。我们还通过生物信息学方法,利用DiscoveryStudio软件展示了SRSF11 K419琥珀酰化的示意图。
值得注意的是,SRSF11的K419琥珀酰化可能参与调控其与下游RNA或其他剪接因子的相互作用。
为进一步验证SRSF11的位点特异性琥珀酰化修饰,本研究采用四种方法。蛋白质印迹分析。结果显示,与THLE2细胞相比, HCC 细胞中SRSF11的琥珀酰化水平显著升高。
一致地,在人类和小鼠原代 HCC 模型中, HCC 组织的SRSF11琥珀酰化水平较非癌性肝(NCL)组织显著增加。随后,在293T细胞中表达了带有Flag标签的野生型SRSF11及其突变体。
实验结果显示,在293T细胞过表达模型中,K419R突变完全消除了琥珀酰化信号。第三,我们制备了特异性识别K419位点琥珀酰化SRSF11的抗体(抗SRSF11Ksu抗体)。
我们进一步验证了上述 HCC 细胞和组织中SRSF11 K419位点的琥珀酰化状态。与预期一致,使用抗SRSF11Ksu抗体检测的K419琥珀酰化结果与泛Ksu抗体检测结果完全吻合。
最后,我们将纯化的Flag-SRSF11与琥珀酰辅酶A共同孵育,结果发现补充琥珀酰辅酶A后重组SRSF11的K419位点琥珀酰化呈现剂量依赖性。
此外,采用抗SRSF11Ksu抗体的免疫组化(IHC)检测进一步显示,与 NCL 组织相比, HCC 组织中SRSF11的K419位点琥珀酰化水平显著升高。这些结果表明SRSF11在 HCC 中K419位点发生琥珀酰化。
为深入探究在K419位点对SRSF11进行琥珀酰化修饰的琥珀酰转移酶,我们在293T细胞中过表达了多种已鉴定的琥珀酰转移酶。实验结果表明,KAT2A是SRSF11 K419修饰的唯一催化酶。
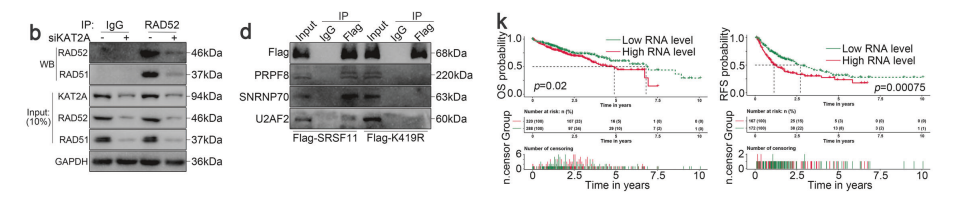
随后,我们分别在LM3和Huh7细胞中过表达和敲低KAT2A。研究发现,过表达KAT2A可增强SRSF11在K419位点的琥珀酰化修饰,而通过siRNA敲低KAT2A则会降低SRSF11在K419位点的琥珀酰化水平。
这些结果表明,K419琥珀酰化可能通过不依赖蛋白质稳定性的机制调控SRSF11的功能。随后,我们通过Co-IP在LM3和Huh7细胞中证实了SRSF11与KAT2A的直接结合。
免疫荧光显示SRSF11与KAT2A在细胞核内共定位。原位邻近连接分析(PLA)进一步支持了SRSF11与KAT2A的相互作用。我们将纯化的Flag-SRSF11与琥珀酰辅酶A在有无KAT2A的条件下进行孵育。结果显示,SRSF11仅在KAT2A存在时才会发生琥珀酰化。
值得注意的是,高剂量辅酶A可抑制KAT2A介导的SRSF11琥珀酰化,这表明辅酶A会与琥珀酰辅酶A竞争结合KAT2A,且琥珀酰辅酶A中的辅酶A基团参与了其与KAT2A的相互作用。
总之,这些数据证实KAT2A能直接在SRSF11的K419位点(HCC)进行琥珀酰化修饰。
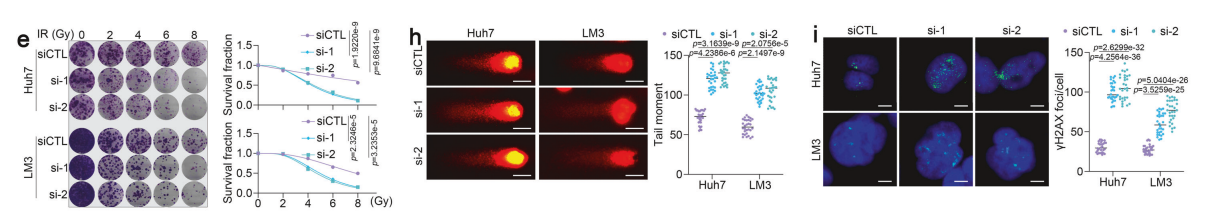
为深入探究SRSF11在 HCC 中的潜在作用,我们通过RNA-seq全面研究了SRSF11的功能。237个失调基因在LM3和Huh7模型中均被观察到。
对这些失调基因进行的通路富集分析(KEGG)显示,沉默的SRSF11与细胞损伤相关信号通路相关,如坏死性凋亡、凋亡和RNA降解。
我们通过siRNA在Huh7和LM3细胞中敲低SRSF11,随后用IR处理细胞。单靶点多靶点模型证实,SRSF11敲低显著抑制了IR处理的 HCC 细胞增殖。
流式细胞术分析进一步证实,SRSF11沉默显著增加了IR诱导的细胞凋亡。G2/M期细胞比例的增加表明细胞对IR更敏感。
我们发现抑制SRSF11可显著增加G2/M期细胞比例。经IR处理的 HCC 细胞中出现DNA断裂。双链断裂(DSBs)是IR诱导细胞死亡的关键机制。
我们通过量化彗星尾矩和磷酸化组蛋白H2AX(γH2AX)焦点来评估IR诱导的细胞DSBs。彗星实验结果显示,与对照组相比,SRSF11沉默的 HCC 细胞在IR处理后表现出更严重的DSBs。
为进一步验证上述结果,我们构建了含有靶向SRSF11的shRNA的慢病毒。抑制SRSF11确实显著促进了电离辐射(IR)诱导的 HCC 细胞死亡和DNA损伤。
此外,通过sgRNA敲除SRSF11也显示出一致结果。重要的是,体内异种移植模型进一步证实了IR后 HCC 中SRSF11敲除的显著协同抑制作用,这通过肿瘤生长和肿瘤重量的结果得以体现。
这些发现确立了SRSF11作为 HCC DNA损伤修复和放射抗性关键调节因子的地位。
接下来,我们进一步探究了SRSF11过表达对DNA损伤和放射敏感性的影响。为此,我们构建了一个含有四环素诱导SRSF11沉默(Tet-off)元件的SRSF11表达载体。
彗星尾矩和 γH2AX 焦点的定量分析进一步表明,SRSF11的过表达可减轻DNA双链断裂(DSBs)。
与细胞实验数据一致,异种移植模型验证了这些发现:SRSF11过表达可消除辐射诱导的肿瘤抑制作用,而TET诱导的SRSF11沉默则恢复了电离辐射(IR)的抑制效果。
基于这些结果,我们提出假说:SRSF11通过调控DNA损伤修复来调节放射抗性。为阐明SRSF11调控DNA损伤修复的潜在机制,我们检测了参与HR和 NHEJ 信号通路的关键基因表达。
结果显示,敲低SRSF11可选择性下调HR介导蛋白RAD51、RAD52、BRCA1和BRCA2的表达,但未改变 NHEJ 组分Ku70/80或 CTIP 的表达。免疫荧光检测证实,SRSF11缺陷细胞在辐照后RAD51核焦点形成减少。
此外,体内 IHC 实验证实,SRSF11沉默后仅HR相关蛋白(而非 NHEJ 蛋白)的表达降低。
相反,SRSF11过表达显著上调RAD51、RAD52、BRCA1和BRCA2的表达,而添加TET后这些因子的表达被重新抑制。
综合这些数据表明,SRSF11通过调控HR而非 NHEJ 促进 HCC 中的DNA双链断裂修复。
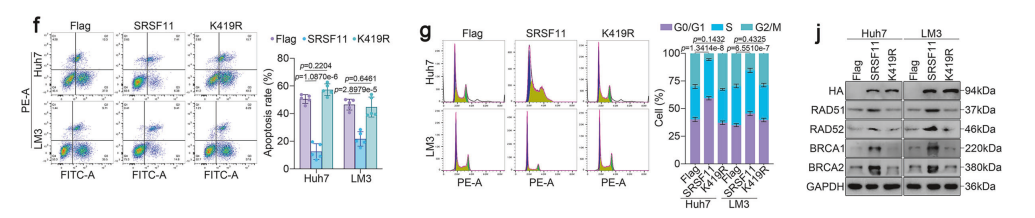
我们进一步探究了KAT2A诱导的SRSF11 K419位点琥珀酰化是否调控 HCC 的同源重组(HR)。
克隆形成实验、细胞凋亡、细胞周期、彗星实验及 γH2AX 焦点分析表明,野生型SRSF11(而非K419R突变体)能显著增强DNA损伤修复能力与放射抗性。
与对照载体相比,野生型SRSF11过表达可明显提升HR相关蛋白的表达水平,而K419R突变体则无此效应。综合数据表明,SRSF11 K419位点对 HCC 的HR过程具有关键调控作用。
为验证KAT2A在 HCC 中催化SRSF11的K419琥珀酰化对人类资源(HR)调控的作用,我们构建了SRSF11基因敲除(SRSF11KO)的Huh7和LM3细胞系。
我们发现KAT2A沉默会导致细胞增殖抑制、促进细胞凋亡、G2/M期阻滞、DNA损伤以及 γH2AX 灶形成。这些效应在K419R重建的细胞中被完全消除。
关键的是,KAT2A敲除选择性调控了野生型而非突变型SRSF11背景下同源重组相关蛋白的表达。
这些数据明确证明,KAT2A介导的K419琥珀酰化使SRSF11在 HCC 中协调同源重组依赖性基因组稳定化的能力得以发挥。
尽管我们已证实K419琥珀酰化能促进SRSF11介导的同源重组(HR),但这些蛋白调控HR的内在机制仍不明确。
我们通过RNA测序技术,在 HCC 细胞中沉默SRSF11以鉴定全局 SRSF 介导的可变剪接事件。
在这些剪接事件中,鉴定出897个差异显著的剪接事件。基因本体(GO)对这些不同剪接基因的分析显示,DNA损伤应答和DNA修复显著富集。
KEGG 分析表明,同源重组(HR)是最显著富集的信号通路,这与我们上述SRSF11调控HR的结果高度一致。
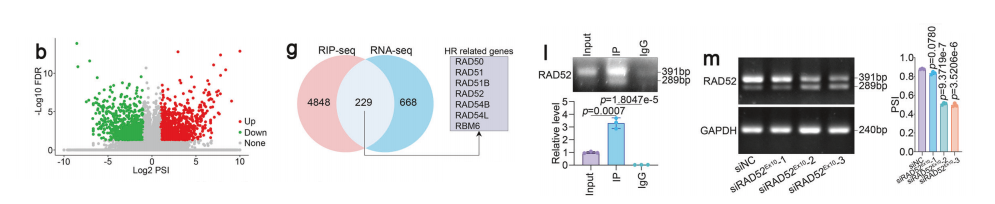
为深入探究SRSF11介导的同源重组(HR)的直接作用靶点,我们采用RIP-seq技术鉴定SRSF11结合的mRNA。通过GO和 KEGG 分析进一步证实,这些基因参与DNA损伤修复和HR过程,信号通路显著富集。
通过对这些SRSF11结合基因及不同剪接事件的整合筛选,我们鉴定出7个同源重组相关候选基因。通过RT-PCR进一步验证其剪接情况,证实仅有RAD51、RAD52、RAD54B和RBM6发生可变剪接。
其中,RAD52的第10号外显子跳跃在SRSF11沉默后显示出显著增加。根据RNA-seq和RIP-seq数据,覆盖度图谱表明SRSF11-IP组中外显子10周围的覆盖度显著增加。
我们使用homer算法鉴定RAD52的SRSF11识别RNA基序,发现含有 CUGAGU 的基序显著富集,该基序位于内含子-外显子连接处附近。
此外,RIP实验进一步验证了SRSF11与RAD52的直接结合。为进一步验证RAD52是SRSF11的直接下游功能靶点,我们设计了特异性靶向RAD52第10外显子的siRNA(siRAD52Ex10)。
实验表明,沉默SRSF11可增加RAD52第10外显子的跳跃。在此基础上,我们进一步证实过表达SRSF11会减少RAD52第10外显子的跳跃。
然而,当通过siRAD52Ex10敲低RAD52表达后,SRSF11的过表达并未显著增加第10外显子的包含。这些数据进一步支持RAD52第10外显子是SRSF11的直接剪接序列。
为明确RAD52外显子10跳跃是否发生于 HCC 患者,我们采用RT-PCR技术检测了 HCC 和 NCL 组织中该基因的表达水平。
在检测样本中, HCC 组织中RAD52外显子10的跳跃现象较 NCL 组织呈现持续性降低。我们设计了特异性靶向外显子10的探针,通过RNA原位杂交技术检测石蜡切片中含/不含外显子10的RAD52表达量。
正如预期,携带外显子10的高表达RAD52与 HCC 患者较差的OS和RFS显著相关。
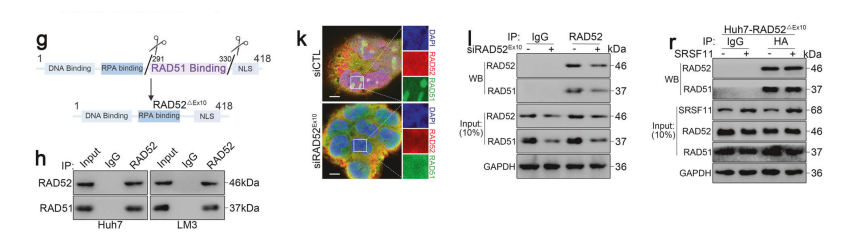
接下来,我们试图探究由SRSF11沉默介导的RAD52第10号外显子跳跃是否参与调控同源重组(HR)。
首先,我们发现SRSF11沉默显著加剧了过表达RAD52的 HCC 细胞中电离辐射(IR)引起的DNA损伤,这一结果通过集落形成实验、彗星实验、 γH2AX 染色(以及有限HR得以证实。
此外,siRAD52Ex10也显著增加了SRSF11过表达 HCC 细胞中IR诱导的DNA损伤和有限HR。
与这些结果一致,在裸鼠异种移植模型中,我们过表达HA-RAD52并敲低SRSF11,发现与对照组相比,敲低SRSF11可显著抑制体内肿瘤生长。
这些结果表明,由SRSF11调控的RAD52第10外显子跳跃可能损害同源重组(HR)。
为进一步验证RAD52第10外显子跳跃对HR的损害作用,我们构建了缺失第10外显子的HA融合RAD52截短突变质粒(HA-RAD52△Ex10)。
敲低SRSF11后,RAD52单独过表达可促进IR诱导的DNA损伤修复,但HA-RAD52△Ex10单独过表达则无法促进DNA损伤修复。这些结果表明,RAD52的第10外显子跳跃抑制了 HCC 中HR介导的DNA损伤修复。
值得注意的是,RAD52外显子10编码的蛋白质(289-322氨基酸)包含促进同源重组(HR)的RAD51结合域(291-330氨基酸)。
RAD51是HR聚合酶介导的模板驱动合成中不可或缺的核心重组酶。因此,我们推断RAD52外显子10的跳跃主要影响RAD52与RAD51在HR中的相互作用。
为验证上述假设,我们研究了RAD52外显子10跳跃对RAD51相互作用的直接影响。首先,我们证实了RAD52与RAD51的内源性双向结合。
随后,通过在Huh7和LM3细胞中过表达HA-RAD52,进一步通过Co-IP实验验证了RAD52与RAD51的结合而过表达HA-RAD52△Ex10后未检测到RAD51与RAD52的显著结合。
免疫荧光检测进一步揭示了 HCC 细胞中RAD52与RAD51的共定位现象。值得注意的是,当使用siRAD52Ex10特异性敲低RAD52转录本的第10外显子后,RAD51与RAD52的结合显著受抑制。
这些结果表明RAD52的第10外显子对其与RAD51的结合至关重要。
为进一步探究SRSF11介导的RAD52第10外显子包含对RAD51结合的影响,我们发现过表达SRSF11能通过免疫共沉淀(Co-IP)促进RAD52与RAD51的结合。
实验结果显示,敲低SRSF11会抑制RAD52和RAD51的结合。与此一致的是,在SRSF11过表达的细胞中,siRAD52Ex10同样导致RAD52和RAD51结合减少。
siSRSF11对RAD52过表达细胞也产生类似影响。然而,在HA-RAD52△Ex10细胞中,SRSF11的过表达对RAD52和RAD51的结合无显著影响。
这些数据证实,SRSF11介导的RAD52外显子10包含许可了RAD51的结合,从而在 HCC 中实现了依赖同源重组的基因组稳定化。
我们随后研究了KAT2A介导的SRSF11 K419位点琥珀酰化在RAD52剪接中的作用。首先证实KAT2A沉默会显著增加RAD52前体mRNA的外显子10跳跃。
同时,KAT2A敲除抑制了RAD52与RAD51的结合。这些结果表明,KAT2A介导的K419琥珀酰化通过SRSF11介导的RAD52外显子10跳跃调控RAD52与RAD51的结合。
接下来,我们进一步探究KAT2A介导的K419琥珀酰化如何调控SRSF11介导的RAD52剪接的潜在机制。我们推测K419琥珀酰化可能影响SRSF11与RAD52前体mRNA及关键剪接蛋白的结合。
为此,我们首先研究了KAT2A介导的K419琥珀酰化是否影响SRSF11与RAD52前体mRNA的结合。
我们在 HCC 细胞中分别过表达Flag-SRSF11或Flag-SRSF11 K419R,发现过表达Flag-SRSF11 K419R的细胞中RAD52前体mRNA与SRSF11的结合显著受抑制。
通过 TCGA 数据库分析,我们检测了剪接体关键蛋白、U2AF2、PRPF4、PRPF8、SNRNP70、SNRPB、 DHX15、BRR2和SF3B1与KAT2A及SRSF11的表达相关性。
TCGA 数据库数据显示,U2AF1、U2AF2、PRPF4、PRPF8、SNRNP70、 SNRPB 、DHX15、SF3B1和BRR2的表达水平在mRNA和蛋白层面均与SRSF11呈正相关。
此外,U2AF1、U2AF2、PRPF4、PRPF8、SNRNP70、 SNRPB 、DHX15、SF3B1和BRR2的表达水平均与KAT2A呈正相关。
这些基因的表达还与RAD51和RAD52的表达呈正相关。由此我们推断,KAT2A介导的K419琥珀酰化作用促进了SRSF11相关剪接体的形成。
为验证该假说,我们通过CoIP证实了SRSF11与U2AF2/PRPF8/SNRNP70复合物的结合。
相反,我们发现过表达SRSF11会促进其与U2AF2/PRPF8/SNRNP70复合体的结合,而敲低SRSF11则会抑制其与该复合体的结合。
值得注意的是,在SRSF11过表达的 HCC 细胞中,敲低KAT2A会抑制SRSF11与这些蛋白的结合,而过表达KAT2A则会产生相反效应。
这些数据揭示了一个KAT2A-SRSF11轴:K419位点的琥珀酰化修饰可激活剪接体募集,从而强制RAD52外显子10的包含,进而提高同源重组效率。
本研究进一步分析了SRSF11表达与HR介导的DSBs修复、放疗敏感性及 HCC 患者生存结局的相关性。
建立了原位小鼠 HCC 模型, IHC 结果表明两种模型中 HCC 组织的SRSF11表达均高于 NCL 组织。
与之一致的是,在我们的临床队列中, HCC 组织的SRSF11表达也显著高于 NCL 组织。来自tcgalihc队列的转录组和蛋白质组数据进一步证实, HCC 组织中SRSF11 mRNA和蛋白水平均显著高于 NCL 组织。
最后,通过单细胞转录组数据(GSE166635),我们进一步验证了恶性细胞中SRSF11表达确实高于非恶性细胞。
我们随后评估了SRSF11对 HCC 患者对激素抵抗(HR)和放疗敏感性的影响。卡方分析显示,SRSF11水平与RAD52(P=0.0001)、RAD51(P=0.0001)、BRCA1(P=0.0222)和BRCA2(P=0.0035)水平呈显著正相关。
与本队列一致, TCGA - LIHC 数据表明SRSF11水平与RAD52、RAD51、BRCA1和BRCA2水平显著正相关。
转录组和蛋白质组数据均表明,高SRSF11表达与 HCC 中HR介导的修复及放射抵抗相关。综上,这些数据有力支持SRSF11赋予HR和放射抵抗的观点。
SRSF11表达与放射抵抗性之间的强关联促使我们探究高SRSF11表达是否与 HCC 患者不良临床结局相关。
确实,我们的队列研究表明,高SRSF11表达与 HCC 患者较差的总生存期(OS)和无复发生存期(RFS)显著相关。
根据 TCGA - LIHC 队列的转录组学和蛋白质组学数据,高SRSF11表达确实与 HCC 患者较短的OS和RFS显著相关。
综合这些结果表明,SRSF11的高表达与患者放射抵抗性和不良生存率相关。
KAT2A/SRSF11抑制所引发的肿瘤细胞死亡——选择性阻断同源重组(HR)而不干扰 NHEJ ——使该通路成为放射肿瘤学中精准治疗的潜在靶点。
我们的研究为联合靶向琥珀酰辅酶A通量和DNA损伤应答通路开辟了新途径,从而增强晚期 HCC 的放疗效果。

作者使用伯信生物明星产品RIP进行分子调控机制研究。
伯信好物推荐
产品介绍:
RNA Immunoprecipitation(RIP)是研究细胞内RNA与蛋白质结合的技术,是了解转录后调控网络动态过程的有力工具,可应用于miRNA调控靶点、RNA 与 RBPs(RNA结合蛋白)互作等研究。

伯信 RIP Kit分为:
Bes5101(S) RIP Kit 12T
Bes5101(N) RIP Kit 40T
实验原理:
RIP技术运用针对目标蛋白的抗体免疫沉淀相应的RNA-蛋白复合物,并纯化与蛋白结合的RNA。最后,结合基因特异性分析技术(PCR、qRT-PCR)或高通量分析技术(高通量测序、基因芯片),分析结合在复合物上的RNA类型及数量。
技术流程:

结果实例:

产品优势:
1. 伯信独立研发。
2. 提供优化的磁珠,适于免疫沉淀RNA-蛋白复合物。
3. 高品质,稳定性好,
4. 结果准确,重复性好。
5. 操作简单,快速省时。

