


Targeting AQP5-mediated arginine deprivation in gastric cancer stem cells restores NK cell anti-tumor immunity
(IF=10.6002)《Cell Reports Medicine》
摘要
自然杀伤(NK)细胞进入肿瘤微环境(TME)后抗肿瘤活性受损,但其具体机制尚不明确。本研究证实,AQP5阳性胃癌干细胞通过重编程尿素循环(UC)导致 NK 细胞功能障碍。
其机制是:AQP5通过竞争性结合核转运蛋白β1亚基(KPNB1)上的ATP依赖性RNA解旋酶A(DHX9),抑制DHX9的核转位,并转录下调精氨酸琥珀酸合成酶1(ASS1)。
在AQP5阳性肿瘤细胞重塑的 TME 中,低精氨酸状态通过限制一氧化氮(NO)合成削弱 NK 细胞功能。值得注意的是,临床前小鼠模型证实,口服精氨酸补充剂可增强 NK 细胞对AQP5高表达胃癌(GC)组织生成类器官的杀伤作用。
此外,AQP5阳性肿瘤细胞还会将尿酸循环(UC)重定向至三羧酸循环(TCA循环),通过促进谷氨酸-氨甲酰胺连接酶(GLUL)稳定性,将储存的氮以谷氨酰胺形式储存。
本研究揭示了AQP5阳性癌症干细胞通过改变自身代谢模式来损害 NK 细胞细胞毒性的证据。
为阐明免疫微环境对AQP5+ GC-CSCs驱动肿瘤进展的贡献,我们比较了免疫功能正常C57BL/6J小鼠与免疫缺陷NSG小鼠(NOD-scid IL2Rγnull)中AQP5+与AQP5−小鼠胃癌细胞(MFC)的肿瘤生长及转移差异。
在两种小鼠模型中,AQP5+ MFC细胞的致瘤性和转移能力均显著高于AQP5− MFC细胞。肺转移结节中也观察到类似趋势,提示免疫微环境可能为AQP5+胃癌进展提供了更有利的微环境。
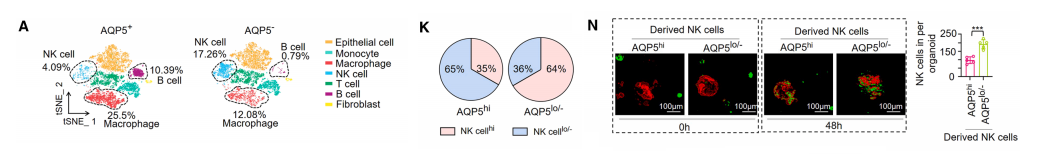
随后,我们对C57BL/6J小鼠皮下肿瘤模型进行了单细胞RNA测序(scRNA-seq),我们发现AQP5+与AQP5-皮下肿瘤中 NK 细胞、B细胞及巨噬细胞簇的比例存在显著差异。
随后,我们对皮下肿瘤中的T细胞、 NK 细胞、B细胞和巨噬细胞进行了分选,以评估AQP5表达是否影响其增殖能力和细胞毒性功能。
与AQP5−肿瘤相比,浸润AQP5+皮下肿瘤的 NK 细胞在增殖和细胞毒性功能上均表现出统计学显著的损害。
为评估AQP5表达是否影响 NK 细胞成熟,我们根据单细胞RNA测序数据将肿瘤浸润的 NK 细胞分为四个不同的成熟亚群。
定量分析显示AQP5+肿瘤中所有亚群的AQP5表达均降低,表明AQP5影响的是 NK 细胞的丰度而非成熟状态。
单细胞RNA测序分析进一步表明,与AQP5−肿瘤相比,AQP5+皮下肿瘤可能诱导 NK 细胞功能障碍,其特征包括细胞毒性效应分子的显著下调、肿瘤归巢能力(Xcl1和Sell)的减弱以及炎症信号传导(Tnf和Il10ra)的受损。
随后,我们对AQP5+GC细胞对 NK 细胞增殖和细胞毒性的影响进行了体外验证。与AQP5−GC细胞相比,从健康供体外周血中分离的原代 NK 细胞与AQP5+GC细胞共培养时,其增殖和细胞毒性活性显著减弱,效应分子表达(IFN - γ 、 TNF - α 和 GZMA)降低。
这些结果表明,AQP5+ TME 可能引发更严重的 NK 细胞功能障碍。
为阐明AQP5在胃癌 NK 细胞功能障碍中的作用,我们对95个胃癌组织微阵列进行了多色免疫荧光分析。仅35%的AQP5高表达组组织表现出高 NK 细胞浸润,这一比例显著低于AQP5低表达组观察到的64%。
这些结果表明,胃癌组织中AQP5的高表达会减少 NK 细胞浸润。此外,我们发现从AQP5高表达胃癌组织中分离的 NK 细胞表现出显著降低的细胞毒性、较低水平的效应分子(IFN - γ 和 TNF - α),以及明显受损的攻击胃癌类器官的能力。
为进一步验证AQP5+胃癌细胞对 NK 细胞介导的抗肿瘤活性的影响,我们在NSG小鼠中建立了异种移植模型和尾静脉注射诱导的肺转移模型。105个 NK 细胞的过继转移显著抑制了AQP5−肿瘤的生长和转移,但对AQP5+肿瘤的影响极小。
这些发现进一步支持了 NK 细胞功能在AQP5+肿瘤背景下受损的观点。
综上,我们的结果表明AQP5+胃癌细胞可能促进了一种免疫抑制 TME ,从而削弱了 NK 细胞的细胞毒性功效。
代谢物在调控细胞活性和生物学行为中起重要作用。为探究胃癌中AQP5表达相关的代谢重编程,我们对AQP5+和AQP5-胃癌细胞进行了非靶向代谢组学分析,揭示了两组细胞间独特的代谢特征。
AQP5+胃癌细胞在多个关键代谢通路中存在显著改变,包括精氨酸生物合成、谷氨酸代谢及中心碳代谢。
为验证这些代谢学发现,我们采用LC-MS/MS定量分析了氨基酸水平。与非靶向代谢组学结果一致,AQP5+胃癌细胞中尿囊素(UC)代谢物(精氨酸和鸟氨酸)水平显著降低,同时谷氨酸和谷氨酰胺浓度升高。
因此,AQP5+胃癌细胞中的UC代谢可能受损。我们通过LC-MS/MS检测了AQP5+和AQP5-胃癌细胞培养基中的这些氨基酸。
值得注意的是,仅AQP5+胃癌细胞培养基中的精氨酸水平降低,而鸟氨酸、谷氨酸和谷氨酰胺与AQP5-胃癌细胞培养基相比无显著变化。
重要的是,在原发性胃癌标本中,精氨酸水平随AQP5表达水平升高(按 IHC 分为四个等级)而显著降低。
综上,这些结果表明胃癌细胞中AQP5的表达可能形成低精氨酸微环境(TME)。
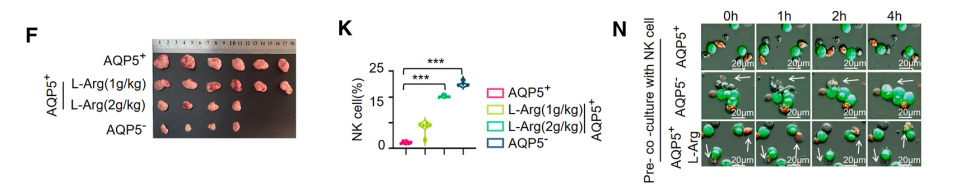
我们随后研究了补充L-精氨酸是否能挽救体内AQP5+胃癌细胞诱导的 NK 细胞功能障碍。通过每日口服灌胃不同剂量L-精氨酸(1-2 g/kg),我们发现L-精氨酸(2 g/kg)显著抑制了AQP5+胃癌肿瘤的发生、进展和转移。
该治疗方案显著增强了 NK 细胞在AQP5+肿瘤中的浸润。重要的是,使用抗NK1.1单克隆抗体清除 NK 细胞后,完全逆转了补充精氨酸的肿瘤抑制效应,证实了 NK 细胞在介导精氨酸治疗效果中的关键作用。
因此,低L-精氨酸条件诱导的 NK 细胞抗肿瘤免疫功能受损,可能是 TME 中AQP5+肿瘤相较于AQP5-肿瘤进展加速的关键驱动因素。
为进一步验证这些发现,我们确认了体外补充L-精氨酸是否能恢复由AQP5+胃癌细胞引起的 NK 细胞功能损伤。
在Transwell实验中,与AQP5−胃癌细胞共培养的 NK 细胞相比,与AQP5+胃癌细胞共培养的 NK 细胞表现出显著降低的肿瘤杀伤能力,而补充L-精氨酸可有效恢复其抗肿瘤活性。
此外,我们发现与AQP5−胃癌细胞共培养的 NK 细胞在Transwell小室中表现出显著低于AQP5+胃癌细胞的细胞内精氨酸水平。
外源性L-精氨酸补充可有效恢复由AQP5+胃癌细胞引起的精氨酸水平耗竭。值得注意的是,外源性L-精氨酸补充显著改善了AQP5+胃癌细胞介导的 NK 细胞增殖、细胞毒性和效应分子表达的损伤。
综上,这些数据提供了有力证据,表明AQP5+胃癌细胞诱导的 TME 中L-精氨酸缺乏是导致 NK 细胞抗肿瘤活性受损的关键机制。
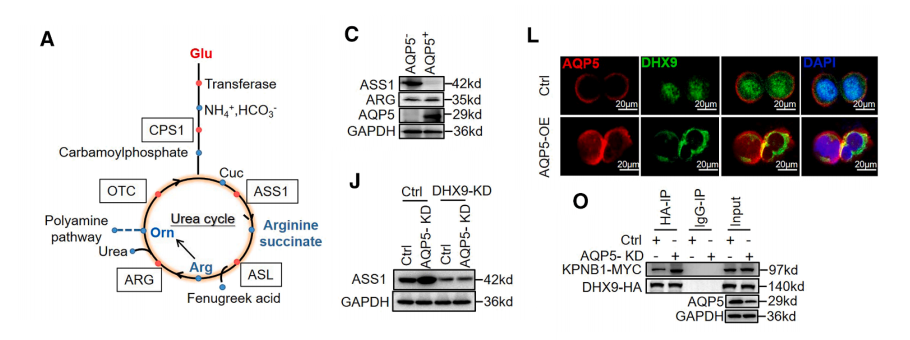
为探究AQP5+胃癌细胞如何建立低L-精氨酸 TME ,我们对关键尿囊素合成酶进行了蛋白质组学分析。
精氨琥珀酸合成酶1(ASS1)在AQP5过表达的胃癌细胞和AQP5+胃癌细胞中均呈现表达下调,而AQP5+胃癌细胞中的精氨酸酶(ARG)水平未受影响。
体内ASS1过表达不仅显著抑制了AQP5+胃癌细胞的皮下肿瘤进展,还增强了 NK 细胞的募集并恢复了 TME 内的L-精氨酸水平。
与此一致的是,AQP5−胃癌细胞中ASS1的缺失促进了体内肿瘤进展,同时伴随瘤内L-精氨酸水平降低和 NK 细胞募集减弱。
综上,这些结果进一步证实了ASS1依赖性精氨酸合成在AQP5+胃癌细胞中调控 NK 细胞抗肿瘤活性的关键作用。临床标本验证进一步表明,与AQP5Low胃癌组织相比,AQP5High胃癌组织中ASS1表达显著下调。
综合这些数据表明,AQP5通过抑制ASS1表达来降低精氨酸的生成。
鉴于AQP5在胃癌(GC)细胞中转录抑制ASS1,我们筛选了转录因子或调控因子,通过Revers-ChIP分析验证了这一效应。
我们筛选出7种转录调控相关蛋白进行进一步验证:DExHBox解旋酶9(DHX9)被确认为ASS1表达的关键调控因子,因为在GC细胞中敲低DHX9可显著逆转AQP5敲低产生的效应——即ASS1上调、L-精氨酸产量增加以及ASS1启动子活性增强。
此外,DHX9在ASS1启动子区域呈现显著富集。DHX9作为转录调控因子通过核转位调控转录活性。我们推测AQP5可能影响DHX9的核转位。
实验结果证实,AQP5过表达会导致DHX9滞留胞质,而AQP5敲低则促进其核转位。因此,AQP5通过抑制DHX9核转位来降低ASS1表达。
我们筛选了与DHX9相互作用的蛋白质,发现KPNB1(核转运蛋白β1亚基)是唯一能与DHX9结合的核转运蛋白。
同时,AQP5显著抑制了DHX9与KPNB1的结合,这一结果通过在AGS和293T细胞系中过表达或敲低AQP5得到进一步验证。
此外,KPNB1与DHX9的相互作用随AQP5表达量增加而逐渐减弱;类似地,AQP5与DHX9的结合也随KPNB1表达量增加而逐渐减弱。这些结果表明AQP5与KPNB1竞争结合DHX9。
特别的是,虽然在对照胃癌细胞中过表达KPNB1促进了DHX9的核转位,但在过表达AQP5的胃癌细胞中,共过表达KPNB1明显逆转了AQP5对DHX9核转位的抑制作用。
这些发现证实AQP5通过竞争KPNB1结合DHX9,从而抑制DHX9的核转位,进而抑制ASS1的转录表达及精氨酸生物合成。
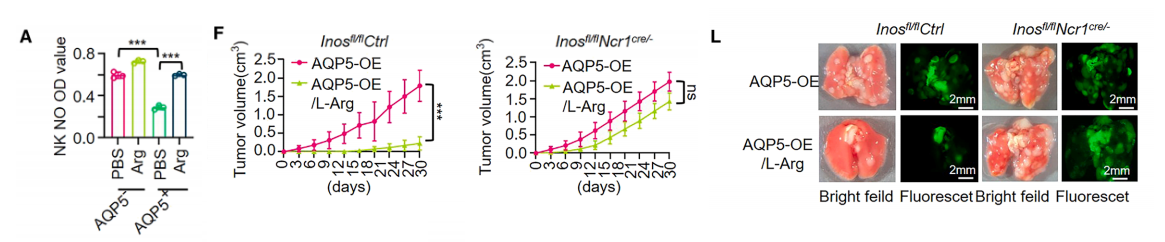
精氨酸是合成多胺、肌酸和脯氨酸的底物,这些物质对细胞存活和增殖至关重要。我们发现,在Transwell实验中与AQP5+ GC细胞共培养的 NK 细胞中,NO水平显著降低,而补充L-精氨酸可恢复NO水平。
三种NOS亚型参与NO合成。药理学抑制iNOS会显著削弱 NK 细胞的NO合成、增殖和细胞毒性,无论AQP5表达水平或外源性L-精氨酸补充情况如何。
值得注意的是,iNOS阻断完全消除了L-精氨酸对与AQP5+ GC细胞共培养的 NK 细胞的增强作用。
此外,从人类胃癌组织中分离的 NK 细胞也显示出相当水平的iNOS表达。这些发现共同表明,iNOS是 NK 细胞中负责精氨酸分解代谢和一氧化氮生成的关键酶。
随后,我们进一步分析了AQP5+ GC细胞产生的低精氨酸 TME 是否通过限制NO合成而损害 NK 细胞的抗肿瘤免疫功能,该研究在 NK 细胞特异性iNOS缺失(Inosfl/flNcr1cre/-)的转基因小鼠中进行。
值得注意的是, NK 细胞条件性iNOS缺失并未影响稳定过表达AQP5的MFC细胞建立的肿瘤发生率或生长。然而,其缺失完全消除了Inosfl/flCtrl小鼠中L-精氨酸补充诱导的肿瘤生长抑制作用。
其次,虽然外源性L-精氨酸补充显著增加了两种小鼠模型皮下肿瘤的瘤内精氨酸水平,但仅显著提高了Inosfl/flCtrl小鼠中肿瘤浸润 NK 细胞和细胞毒性 NK 细胞(TNF - α +或 IFN - γ +)的比例;这种增强作用在Inosfl/flNcr1cre/-小鼠中未观察到。
与这些发现一致的是,L-精氨酸补充未能抑制AQP5过表达MFC细胞在Inosfl/flNcr1cre/-小鼠中的肺转移,但在Inosfl/flCtrl小鼠中显著降低了转移负荷。
这些数据表明,由AQP5+胃癌细胞引起的 TME L-精氨酸缺乏会通过限制一氧化氮(NO)合成,严重损害 NK 细胞介导的肿瘤免疫。
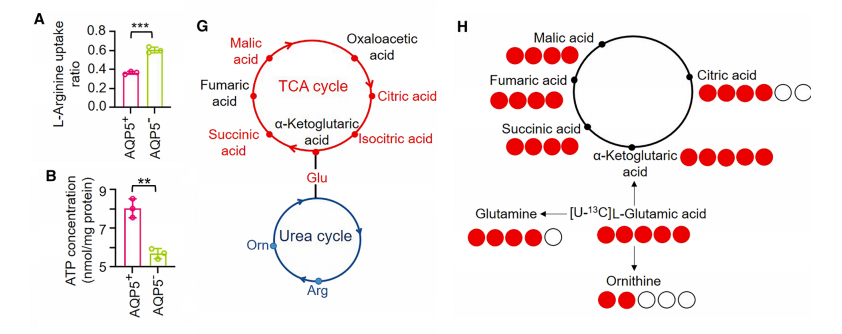
精氨酸可影响细胞生物能量学,包括ATP水平、ADP/ATP比值及氧消耗率(OCRs)。我们观察到L-精氨酸摄取显著降低与AQP5– GC细胞相比,AQP5+ GC细胞。
为确定L-精氨酸缺乏是否影响AQP5+ GC细胞的生物能量学,我们进行了生物能量学分析。出乎意料的是,AQP5+ GC细胞表现出ATP水平和OCRs升高。
此外,AQP5+ GC细胞中ASS1的过表达逆转了这种生物能量学表型,表明AQP5+ GC细胞通过激活替代代谢途径来补偿L-精氨酸缺乏。
我们推测AQP5阳性胃癌细胞会将氮代谢物重新导向TCA循环以满足其增强的生物能量需求。在AQP5阳性胃癌细胞和AQP5高表达肿瘤组织中均观察到谷氨酸水平升高。
靶向代谢组学分析显示,AQP5阳性胃癌细胞中TCA循环中间产物显著积累。这些结果表明,在这些细胞中,增加的谷氨酸进入TCA循环而非尿酸循环。
为明确AQP5表达对胃癌细胞谷氨酸代谢的影响,我们对AQP5+和AQP5-胃癌细胞进行了[U-13C6]谷氨酸示踪实验,并通过质谱分析进行验证。
结果显示,AQP5+胃癌细胞中M+4琥珀酸、M+4延胡索酸和M+4谷氨酰胺的含量显著增加,同时M+2鸟氨酸的富集程度降低。
这些发现表明,AQP5+胃癌细胞通过将谷氨酸导向谷氨酰胺和三羧酸循环中间产物,同时减少尿酸通量。这种代谢重编程使AQP5+胃癌细胞在维持生物能量代谢的同时,通过增强三羧酸循环实现了氮流失的最小化。
谷氨酰胺代谢在细胞生物能量学中起着关键作用,我们的分析显示,AQP5+ GC细胞和AQP5High GC组织中的谷氨酰胺水平显著升高。[U-13C6]谷氨酸示踪显示,AQP5+ GC细胞中的谷氨酰胺合成通量增强。
这些发现表明,AQP5+ GC细胞通过抑制尿素循环以最小化氮损失,从而优化其生物能量学能力,进行代谢重编程以积累谷氨酰胺作为三羧酸循环的能量储备。
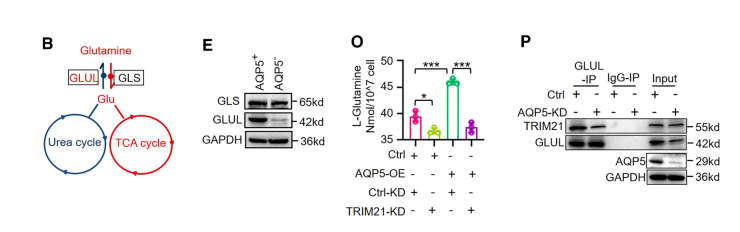
随后,我们通过小干扰RNA(siRNA)沉默 GLUL 和GLS。GLS沉默导致谷氨酰胺随时间积累并伴随ATP逐渐减少,表明谷氨酰胺分解代谢通过三羧酸循环输入对维持细胞生物能量学至关重要。
相比之下, GLUL 沉默表现出明显的时间效应:24小时沉默仅部分降低谷氨酰胺水平(仍高于AQP5−对照组),且不影响ATP生成;而延长至48-72小时的沉默则使谷氨酰胺降至与AQP5- GC细胞相当的水平,并显著降低ATP含量。
基线表达分析显示AQP5+ GC细胞中 GLUL 表达显著高于AQP5− GC细胞,而GLS水平相当。关键的是,沉默 GLUL 48小时不仅降低谷氨酰胺水平,还逆转了AQP5+ GC细胞中观察到的L-精氨酸减少,并显著抑制AQP5+ GC细胞的增殖和迁移。
这些结果表明,AQP5+ GC细胞通过 GLUL 上调维持谷氨酰胺合成以支持三羧酸循环,其机制是将氮通量从尿囊液(UC)转向谷氨酰胺积累。
为阐明AQP5如何调控 GLUL 表达,我们进行了全面的分子分析。有趣的是,虽然在GC细胞中过表达AQP5并未改变 GLUL 转录水平,但显著增强了 GLUL 蛋白的稳定性。
我们检测了 GLUL 的泛素化模式,发现AQP5过表达增加了 GLUL 的泛素化。AQP5过表达特异性促进了 GLUL 的K63连接泛素化,而不影响K27或K48连接的泛素化。
这一发现与我们先前的研究结果一致,即与经典的K48连接泛素化不同。为进一步验证这一机制,我们采用遗传互补方法:在耗尽内源性泛素后,用野生型血凝素(HA)标签泛素重建细胞。
(K63-Ub)或K63突变泛素(K63R)。值得注意的是,K63突变完全消除了AQP5介导的 GLUL 蛋白稳定化。
这些结果表明,AQP5通过特异性K63连接泛素化促进 GLUL 蛋白稳定性,揭示了糖皮质激素代谢中一种先前未被认识的翻译后调控机制。
为阐明AQP5介导 GLUL 泛素化的机制,我们进行了基于质谱的相互作用组分析。在所有鉴定的 GLUL 相互作用蛋白中,TRIM21成为唯一与泛素化密切相关的E3泛素连接酶。
我们的免疫共沉淀实验证实存在由AQP5、TRIM21和 GLUL 组成的三元复合物。令人惊讶的是,TRIM21在293T细胞中显著促进了而非降低了 GLUL 蛋白稳定性。
因此,我们假设AQP5通过TRIM21诱导 GLUL 泛素化。此外,TRIM21敲除不仅降低了 GLUL 的K63连接泛素化水平和谷氨酰胺水平,还消除了AQP5诱导的 GLUL K63连接泛素化及谷氨酰胺水平升高。
由此我们推测,AQP5可能通过促进 GLUL -TRIM21相互作用来增强 GLUL 蛋白稳定性。
与我们的假设一致,AQP5的过表达显著促进了 GLUL 与TRIM21的相互作用,而AQP5敲除则减弱了它们的结合。
为进一步验证AQP5调控GLULTRIM21相互作用的作用,我们在293T细胞中进行了AQP5的梯度过表达实验。
结果显示,梯度过表达AQP5可逐步增强TRIM21- GLUL 相互作用、 GLUL 的K63连接泛素化以及随之而来的 GLUL 蛋白稳定化。
这些数据表明,AQP5通过将E3连接酶TRIM21募集至 GLUL ,从而促进 GLUL 的K63连接泛素化,进而维持 GLUL 蛋白的稳定性。
与既往发现一致,与AQP5−细胞相比,AQP5+胃癌细胞表现出更强的LC3I向LC3II转化,提示其自噬活性增强。ULK1抑制剂在结直肠癌和胰腺癌等多种肿瘤中已显示出初步临床疗效,但其治疗获益仅限于部分患者。
本研究发现,AQP5通过减少精氨酸生成损害 NK 细胞活性。我们提出假说:AQP5通过ULK1介导的肿瘤细胞自噬激活及 NK 细胞细胞毒性的削弱,共同发挥促肿瘤作用。
因此,将ULK1抑制剂与外源性L-精氨酸联合应用,可能成为AQP5高表达胃癌的协同治疗策略。为验证该策略,我们采用患者来源类器官(PDOs)和患者来源异种移植(PDOX)模型,评估ULK1抑制单独或联合L-精氨酸补充的治疗效果。
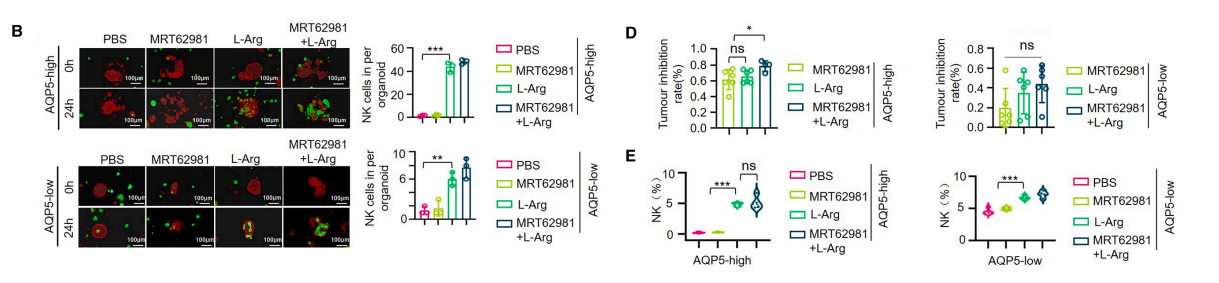
在将 NK -92细胞与PDOs共培养于Matrigel的实验中,我们发现L-精氨酸处理能显著促进 NK -92细胞分化为AQP5高表达PDOs,而L-精氨酸与ULK1抑制剂联用则会严重破坏其结构完整性。但无论是单独使用ULK1抑制剂还是L-精氨酸,都无法完全瓦解类器官结构。
值得注意的是,与AQP5高表达PDOs相比,这种联用效应在AQP5低表达PDOs中明显减弱。这些结果表明,该联合策略在体外 PDO 模型中对AQP5阳性胃癌具有更优的治疗效果。
为进一步验证我们的联合治疗策略,我们使用在NV-NSG-hIL15小鼠中重建功能性人类免疫系统的外周血单个核细胞(PBMCs)构建了AQP5高表达或低表达的胃癌组织 PDOX 模型。
初步表征证实皮下肿瘤中均存在AQP5表达水平,且外周血中 NK 细胞成功重建。ULK1抑制剂与L-精氨酸的联合治疗在AQP5高表达 PDOX 模型中显示出显著的肿瘤消退效果,而在AQP5低表达模型中效果微弱。
值得注意的是,与AQP5低表达肿瘤相比,L-精氨酸单药治疗对AQP5高表达肿瘤表现出更强的疗效。与体外 PDO 模型结果一致,L-精氨酸治疗还促进了 NK 细胞向肿瘤的浸润,尤其在AQP5高表达肿瘤中。
这些结果共同表明,ULK1抑制与L-精氨酸补充的协同联合治疗策略,为AQP5高表达的胃癌患者提供了一种具有前景的治疗方案。
本研究结果表明,AQP5+癌症干细胞的特异性代谢编程不仅能抑制有效抗肿瘤免疫应答的产生,还可为自身储备能量。
值得注意的是,我们证实 TME 中精氨酸的缺乏会严重损害 NK 细胞功能,这为开发更优的 NK 细胞免疫治疗策略提供了理论依据。

作者使用伯信生物明星产品Reverse-ChIP进行分子调控机制研究。
伯信好物分享
Reverse-ChIP Kit
产品介绍:
反向染色质免疫共沉淀技术(reverse chromatin immunoprecipitation assay,Reverse ChIP)相对应于染色质免疫共沉淀技术(ChIP),是一种在体内状态下分析 DNA-蛋白质相互作用的新方法。
该方法可对靶 DNA 位点相关蛋白质进行全面、系统的鉴定,特别是寻找已知DNA 元件相应的调控蛋白。在发现、鉴定未知调控蛋白和研究 DNA-蛋白质相互作用中有重要应用价值。除细胞外,Reverse ChIP也可应用于微生物、植物等样本。

伯信 Reverse-ChIP Kit分为:
Bes5005(S) Reverse ChIP 12T
Bes5005(N) Reverse ChIP 30T
实验原理:
将细胞内的蛋白质和DNA交联后,利用超声波将已交联的DNA随机切断为一定长度范围内的染色质小片段,然后用特异的核酸探针捕获靶DNA片段和与其结合的蛋白质,最后,用Western Blot或质谱检测和鉴定结合于靶DNA位点的全部相关蛋白质。
技术流程:

结果实例:

产品优势:
1.伯信独立研发。
2.快速省时,整个实验过程不到6小时。
3.可重复性好,特异性强,灵敏度高。
4.操作简便、结果准确可靠。

